
Abstract
Novel coronavirus disease 2019 (COVID-19) emerges as a serious threat to public health globally. The rapid spreading of COVID-19, caused by severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2), proclaimed the multitude of applied research needed not only to save the human health but also for the environmental safety. As per the recent World Health Organization reports, the novel corona virus may never be wiped out completely from the world. In this connection, the inhibitors already designed against different targets of previous human coronavirus (HCoV) infections will be a great starting point for further optimization. Pinpointing biochemical events censorious to the HCoV lifecycle has provided two proteases: a papain-like protease (PLpro) and a 3C-like protease (3CLpro) enzyme essential for viral replication. In this study, naphthyl derivatives inhibiting PLpro enzyme were subjected to robust molecular modelling approaches to understand different structural fingerprints important for the inhibition. Here, we cover two main aspects such as (a) exploration of naphthyl derivatives by classification QSAR analyses to find important fingerprints that module the SARS-CoV PLpro inhibition and (b) implications of naphthyl derivatives against SARS-CoV-2 PLpro enzyme through detailed ligand–receptor interaction analysis. The modelling insights will help in the speedy design of potent broad spectrum PLpro inhibitors against infectious SARS-CoV and SARS-CoV-2 in the future.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is just alarming and ever surprising how rapidly a communicable novel coronavius (2019-nCoV), cause of novel coronavirus disease 2019 (COVID-19), has been spreading in the world in the twenty-first century [1,2,3]. 2019-nCoV is also known as severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2). World Health Organization (WHO) characterized COVID-19 as world pandemic [4]. So far, more than million deaths have been reported from 216 countries and territories [5]. Every death makes us painfully aware that our swords are blunt till date in the battle against this hazardous COVID-19.
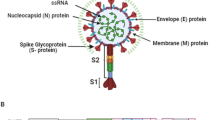
Researchers around the world have been trying different options to restrict the virus replications [6,7,8,9,10,11,12,13,14,15]. However, there is still no effective drug/vaccine against this virus. In this situation, emphasis should also be given to the systematic rational drug discovery against different targets of the virus. Among the different targets, two proteases namely papain-like protease (PLpro) and a 3C-like protease (3CLpro) are very crucial for virus replication and are considered as important druggable targets [3, 6, 7, 16,17,18,19,20,21,22]. The PLpro enzyme also shows deubiquitinating (DUB) and deISGylating activities [9, 16]. As a result, it is also responsible for host cell immune suppression due to the inactivation of NF-κB pathway (Fig. 1). In addition, the structures of different PLpro enzymes are very similar in different coronavirus, and therefore, it is considered as a target for broad-spectrum inhibitor development.

Schematic plot for SARS-CoV-2 genome (PLpro enzyme is highlighted in red color)
Naphthalene is the most straightforward member of the class of PLpro inhibitors [23,24,25,26], in which a couple of benzene rings are fused in the ortho positions. Numerous naphthalene-containing molecules have also been reported to boast significant antimicrobial property. A commonly used dye, β-naphthol, exhibits antimicrobial activity [27]. In addition, naphthyl-based drugs including naftifine, terbinafine, nafacillin, tolnaftate, etc., are found to possess antimicrobial property [27,28,29]. Ratia and collaborators first introduced naphthyl derivatives those were likely to act as non-covalent competitive inhibitors of PLpro [23]. Naphthyl derivative binds within the S4-S3 subsites of the enzyme, thereby inducing a loop closure which ultimately results in conformational change and manifests the PLpro active site as non-functional. Several other naphthyl derivatives were further reported as PLpro inhibitors [23,24,25,26]. In recent times, the interest on naphthyl derivatives is tremendously increased as these derivatives have shown potential SARS-CoV-2 PLpro inhibition [30].
Thus, in this work, we have focused our attention on naphthyl derivatives as SARS-CoV-2 PLpro inhibitors. Multiple modelling strategies were applied with these mottos: (a) identification of important fingerprints that modulate the SARS-CoV PLpro inhibition and (b) scope of naphthyl derivatives to target SARS-CoV-2 PLpro though ligand–receptor interaction analysis. The current study, a part of our rational antiviral drug design and discovery programme [31,32,33,34], may offer an initiative to explore the possibility of broad spectrum inhibitors against the PLpro enzyme in case of both SARS-CoV and SARS-CoV-2.
Methods and materials
Dataset
In order to accelerate the drug discovery effort against coronavirus, a set of diverse naphthyl derivatives belonging to a collection of SARS-CoV PLpro inhibitors were collected with inhibitory activities (IC50) [23,24,25,26]. The naphthyl derivatives with no activity and without definite activity were eliminated initially and followed by removal of duplicates from the study. Thus, remaining fifty-six molecules were considered for the further study (Table 1).
Classification-based QSAR study
The classification modelling assists to classify the active and inactive molecules in terms of their biological data. Here, we have employed structural and physico-chemical interpretation (SPCI) [35, 36] and Monte Carlo-based coral QSAR [37,38,39,40,41] studies.
For these studies, the SARS-CoV PLpro pIC50 value, 5.3, was considered as the threshold value. Compounds having the SARS-CoV PLpro pIC50 value of 5.3 or more were classified as higher PLpro inhibitors or actives and those with less PLpro pIC50 than threshold value were distinguished as inactives.
Structural and physico-chemical interpretation (SPCI) analysis
Structural and physico-chemical interpretation (SPCI) study [35, 36] was solicited to identify and estimate contributions of scaffolds and/or linkers and/or single substituent to these naphthyl-based SARS-CoV PLpro inhibitors. At first, descriptors were calculated by the aid of SiRMS tool followed by model development and validation. Four different classification QSAR models were undertaken by using machine learning approaches including gradient boosting classification (GBC), random forest (RF), support vector machine (SVM), and k-nearest neighbour (kNN). The developed models were evaluated by statistical parameters such as balanced accuracy, sensitivity, and specificity [36]. Furthermore, molecular fragmentation of the dataset was done to estimate the contribution from the developed models. Fragments consist of at most three attachment points were preferred, and subsequently, preferred fragments were counted by RDKit in combination with SMARTS pattern [! #1]! @! = ! # [! #1]. Lastly, the overall contribution of the different fragments and their median fragment contribution graphs were generated by using rspciR software package [42].
Monte Carlo optimization-based QSAR study
Monte Carlo optimization method was used to identify the important structural fingerprints that are exclusively responsible for promoting or hindering of activity [37,38,39,40,41]. Here, the molecular structures of the different inhibitors were represented by SMILES (Simplified Molecular Input Line System) format. These symbolic notations were used to represent structural attributes such as atoms, bonds, etc., and to calculate different molecular optimal descriptors (DCW) used for QSAR modelling study.
In this method, the models were generated on the basis of three optimal descriptors like: SMILES, graph and hybrid. The SMILES-based optimal descriptors used in the study were calculated as:
where T represents threshold and N is number of epoch used for generation of models. Further, the constants like: a, b, c, d, α, β and γ represents different coefficients that are used to modify the descriptors. The CW stands for correlation weight of specific structural attributes for PLpro inhibition.
Global SMILE attributes include NOSP (presence or absence of nitrogen, oxygen, sulphur and phosphorus atom), HALO (presence or absence of halogen groups like: fluorine, chlorine, bromine and iodine), BOND (presence or absence of bond like: = is double bond, # is triple bond and @ is stereo-chemical bond), and ATOMPAIR (represents presence of two atom consecutively). The local SMILE attributes are Sk (signifies presence of only one SMILES atom like: C……, N….. etc.), SSk (combination of two SMILES atoms like: C….N…., C….C…. etc.), and SSSk (denotes combination of three SMILES like: C…..N…C, N…(….c…. etc.).
The graph-based descriptors comprise of three types they are: GAO (graph of atomic orbital), HSG (hydrogen-suppressed graph) and HFG (hydrogen-filled graph). These graph-based descriptors are calculated by means of different molecular connectivity indices like: 0ECk,1ECk and 3ECk. The graph-based descriptors are represented as:
Here, the chemical atoms like C, N, O, etc., are represented by Ak. The coefficients having value 0 and 1 are denoted by as α, β and γ. The coefficients having value 1 are generally used for model building, whereas 0 value are excluded. Further, the notations like: 0ECk, 1ECk and 3ECk denotes different Morgan’s connectivity indices used in the study [41]. Thus, the combination of SMILES and graph-based descriptors forms hybrid descriptors which are represented as:
In our study, twenty-one robust classification models were generated from three different splits on the basis of optimum descriptors (SMILES, graph and hybrid) using the balance of correlation method. The dataset consisting of 56 compounds was distributed into the sub-training (25 compounds), calibration (21 compounds) and test (10 compounds). The training set compounds were used for model building, calibration set was used to check the predictive potential and to prevent overtraining of the model, and the test set is used as an estimator to validate the models developed using training and calibration set [43, 44]. Optimization of T (threshold) and N (epoch) values are also performed separately for each model. Here, the number of threshold required for molecular feature extraction from SMILES is represented by T value, and N (epoch) value denotes number of iterations used in Monte Carlo optimization methods (Fig. 2).

Schematic representation of current work design
Molecular docking study
The molecular docking experiments were implemented with the help of AutoDock Vina [45]. Prior to docking study, polar hydrogen atoms were added by the aid of AutoDock Tools (ADT) [46] in order to relax the conformational strain. The docked poses of naphthyl derivatives were visualized by using the Discovery Studio 3.5 Visualizer [47].
MD simulations
The MD simulations for apo and each complex form of the protein were conducted by the GROMACS v5.1.4 [48] using GROMOS96 45a3 force field [49] for the parameterization of the protein. The ligand topology was generated through GROMOS96 force field by the PRODRG 2 server [50]. Each of the system was solved by the SPC/E water molecules, and the charge on simulating systems was neutralized by providing the equal number of counter ions [51]. Then, minimization of the systems was executed by the steepest descent algorithm in 50,000 steps. After the minimization, each system was subjected to the position restrained NVT (300 K) and NPT (1 atm) simulations of 100 ps for the equilibration according to the mentioned temperature and pressure. Equilibrated systems were then submitted for the non-position restrained production simulations of 20 ns each, and the generated data of MD were then used for the enumeration of the RMSD, RMSF and Rg data of the protein backbone, which helps in structural analysis during dynamics. The binding energy of each complex was calculated by g_MMPBSA package of the GROMACS considering 200 frames from 20 ns data of simulations [52].
Result and discussions
In this twenty-first century, health crisis posted by SARS-CoV-2 outbreak, drug repurposing and/or screening of SARS-CoV inhibitor databases is the fastest option in terms of strategic and economic way. However, systematic and rational drug discovery approaches to find potent inhibitors against different targets of SARS-CoV-2 should not be ignored. In this connection, the inhibitors already designed against SARS-CoV will be very helpful due to the sequence similarity between them. Different modelling approaches were applied on naphthyl derivatives as SARS-CoV PLpro inhibitors as outlined in Fig. 2.
Classification-based QSAR study
Performing classification-based structural and physico-chemical interpretation (SPCI) and Monte Carlo-based coral QSAR methods enable interesting visualization of important chemical sub-structural features attributed to enhance or decrease the SARS-CoV PLpro inhibitory properties. Considering the potency threshold value, out of 56 compounds, 31 compounds were identified as lower and 25 compounds were denoted as higher SARS-CoV PLpro inhibitors.
SPCI analysis
In our study, four different models like gradient boosting machine (GBM), random forest (RF), support vector machine (SVM) and k-nearest neighbour (kNN) were developed. The different tuning parameters used to build the machine learning models for naphthyl-based PLpro inhibitors are given in Table S1. The models generated by using SPCI analysis were found to have acceptable statistical parameters as shown in Table 2.
Among four different models (GBM, RF, SVM and kNN), GBM and RF models are found to have similar statistical parameters. Further, a consensus model was generated in order to remove the biasness of the individual model; this was further considered for the interpretation. The contributions of different fragments obtained from the SPCI analysis are shown in Fig. 3.

Contribution plot of different fragments (present in at least 5 compounds) identified by using consensus (red), GBM (light green), kNN (dark green), RF (cyan), and SVM (purple) models. The numbers M and N are different, signify the number of compounds containing a fragment and the number of fragments present in the dataset, respectively as some compounds have several identical fragments and contributions of those fragments were calculated separately
A limpid trend of structure–activity (SARS-CoV PLpro inhibition) relationship is recognized in Fig. 3. Piperidine moiety, acyclic CONH, aromatic NH2, aromatic halogen and phenyl unsubstituted functions exhibit positive effects on SARS-CoV PLpro inhibitory activities, whereas phenoxy and methoxy group attached with an aromatic ring suggest clear negative influence on biological properties (Fig. 3).
Monte Carlo-based Coral QSAR study
In Monte Carlo optimization, a total of twenty-one different models from three different splits were generated using SMILES and graph-based descriptors with a combination of different connectivity indices calculated for generation of different models (Table S2). Each models were developed after the search for desirable T (threshold) and N (epoch) values. Among these models (Table S2), the model M3 (SMILES and GAO with 1ECkconnectivity of split-1) was found to be the best model and was used for the physical interpretation of structural fingerprints for PLpro inhibition (Table 3).
The best model from each split is shown in bold face.
The end point values calculated for M3 are shown below:
Different structural attributes of the best model M3 obtained from Monte Carlo optimization method are depicted in Table S3.
Interpretation of the QSAR models
Each compound of this dataset can be represented as consisting of three parts: head and tail connected by a linker moiety (Fig. 4).

Naphthyl derivaties featuring head (cyan) and tail (purple) connected by a linker marked as red
As the head region is structurally conserved in between 1 and 2-naphthyl, maximum influenced sub-structure/fingerprints were noticed from linker and tail portions. More interestingly, a limpid trend of structure–activity (SARS-CoV PLpro inhibition) relationship was recognized in Fig. 3. From the SAR study, it can be conferred that the 1-naphthyl head was preferable over the 2-naphthyl prototypes. Thus, the polarizability of the 1-naphthyl function modulates the binding against SARS-CoV PLpro (Fig. 5a). The importance of a single methyl substituent at the R3 position when compared to unsubstituted compounds 43–45, 55–56. Notably, the stereo-chemical pattern of the methyl substituent was a critical factor to modulate PLpro binding affinity. This can be understood by observing the X-ray crystal structure of the compound 53-bounded SARS-CoV PLpro complex (Fig. 5b) where the (R)-methyl enantiomer extends into an interior of the PLpro enzyme between Tyr-265 and Thr-302.24 Similarly, (R)-1-naphthylethylamide of compound 9 interacted with Tyr-265 and Tyr-269 aromatic rings and with side chains of Pro-248 and Pro-249 and consequently forms hydrophobic interactions.23 Báez-Santos and collaborators24 very nicely explained the loss of PLpro inhibitory activity with proportional to substituent size at R3 position, due to the potential entropic gain by displacing the water molecules lead to a larger enthalpic penalty of breaking the H-bonds (between these water molecules and aminoacid residues D165, R167, Y274, T302 and D303). Thus, in all docking calculations as well as reported crystal structures with different inhibitors (PDB: 4OW0, 3MJ5, 4OVJ), the conserved water molecules were present in the binding site for ligand–receptor interactions.

Importance of different fingerprints for the inhibition of SARS-CoV PLpro enzyme: Orientation of 1-napthyl group in interactions with P248, P249 as well as Y269 for the best active inhibitor 33 in PDB: 4OW0 (a); position of methyl substituent of inhibitor 53 in interaction with T302 and Y265 in PDB: 3MJ5 (b); position of unsubstituted phenyl moiety in the docking pose of inhibitor 22 (c); interaction of piperidine moiety in the linker part with the amino acids in PDB: 4OW0 (d); Importance of CONH fragment in H-bond interactions with the main chain of amino acids G164, Y269 in PDB: 4OW0 (e); interaction of amino group in the docking pose of inhibitor 77 with the amino acid D165 (f)
Piperidine moiety, acyclic CONH, NH2, aromatic halogen and phenyl unsubstituted functions exhibited positive effects on SARS-CoV PLpro inhibitory activities (Fig. 5c–f). However, the methoxy group attached with an aromatic ring suggested clear negative influence on biological properties. Figure 3 predicted the positive contribution of acyclic CONH and aromatic NH2 in the PLpro inhibition. This can be justified by the SAR observation. At the middle position, the amide NH was demonstrated to be an optimal feature for PLpro inhibition. Addition of a methyl group at amide nitrogen resulted in significantly differing activity levels (the N-methyl derivative 67, IC50 = 22.6 μM vs compound 6, IC50 = 2.3 μM). In addition, the aromatic NH2 was found to be conducive for compounds 9 (IC50 = 0.6 μM), 29 (IC50 = 11.1 μM), 7 (IC50 = 2.6 μM), 40 (IC50 = 2.7 μM), 26 (IC50 = 5.7 μM), 27 (IC50 = 0.39 μM); consequently, this feature increased the biological property against SARA-CoV PLpro. These observations were also in agreement with the descriptors of Monte Carlo-based QSAR analysis where the structural attributes ‘N…1…(…’ explained importance of a nitrogen atom outside any ring which is attached to an aromatic ring having branching (Table S3).
Table S3 demonstrated the importance of ‘C…(…1…’ that can be decoded as sp3 carbons with branching attached to a ring. This is explained by the presence of methylamine fragments, another promoter of activity which contributes to increase of biological activity against SARS-CoV PLpro. This can be explained by compound 77 where incorporation of 5-methylamine substituent on the benzamide ring would result in escalation of the inhibitory activity (IC50 = 0.46 μM). Thus, addition of donor groups (− NH2) at the benzamide ring can lead to promising PLpro inhibitory activity (Fig. 5f). Further introduction of a methyl function to the amine group of 77 yielded compound 78 with minutely narrowed PLpro inhibitory activity (IC50 = 1.3 μM). However, compound 78 exhibits slight enhancement of anti-SARS-CoV potency (EC50 = 5.2 μM) compared to compound 77 (EC50 = 6 μM). Compounds 65 and 68 were poor PLpro inhibitors, both comprising p-NH2 at R2 position. Therefore, it was evidenced that p-NH2 alone at R2 position might hamper the binding with the PLpro enzyme.
The presence of ‘O…(…1…’ (as evidenced by the Monte Carlo-based QSAR analysis) can be decoded by comparing compounds 43–45, 50, 61 and 63 where OCH3 substitution negatively influenced the PLpro inhibitory activity (IC50 range 30 to 149 μM). Thus, increasing the level of negatively charged atom with hydrophobic characteristics of methoxy function at the terminal phenyl ring would result in fall of the biological activity against PLpro. This observation can be understood by the molecular interaction that substituents in 4-position of tail phenyl ring should be from a positively charged group with additional demand of hydrophilic effects.
As mentioned previously, Fig. 3 showed the contributions of the unsubstituted phenyl function, positively, to the SARS-CoV PLpro inhibitory activity (Fig. 5c). This observation may clearly be explained by comparing the naphthyl derivatives 18–22 (bearing unsubstituted phenyl tail), whose IC50 values were found in between the range of 1.9 and 18. However, the marked differences in the PLpro inhibitory activities of these compounds were due to the type of R4 substituents. Moreover, the effect of stereochemistry of R4 substituents ‘C…@…….’ (as evidenced by the Monte Carlo-based QSAR analysis) played critically (compounds 21 vs 22). A tenfold decrease in PLpro inhibitory activity was manifested for (R)-methoxymethyl containing 21 (IC50 = 18 μM), compared to the corresponding S-isomer 22 (IC50 = 1.9 μM). Overall, the minute observation of the SAR study of these naphthyl derivatives 18–22 suggested that R4 substituents were not at all pleasurable towards biological activity because the most active analogue in this series, 22, was similar effective the R4 unsubstituted prototype 18 (IC50 = 2.2 μM).
More interestingly, methylcarboxamide (compounds 24–25) and more electronegative halogen (compounds 32–33) substitutions at the 3rd or 4th position of the terminal phenyl ring manifested improvement in potency compare to the corresponding unsubstituted analogue 18. Figure 3 clearly expressed the importance of aromatic halogen substitutions.
Additionally, dioxolane derivatives 53–54 and 4-ethyl prototype 23 (IC50 = 0.47 μM) also showed promising inhibitory activity against PLpro. The dioxolane group contributed positively to the PLpro inhibitory property not because of the interaction with Gln270 only; there should be another effect which contributed largely to the substituted phenyl derivatives to improve the biological potency. As noted, the mono-fluoro substitution at the phenyl ring induced conspicuous polarization effects in the π-system of the associated terminal phenyl ring and consequently, showed improved binding affinity with the PLpro active site amino acids residues.
Notably, the substitutes at the phenyl ring ‘[…(…1…’ (as evidenced by the Monte Carlo based QSAR analysis) were sensitive to the positional isomers (meta vs para trail). The PLpro inhibitory potency of the compounds bearing the acetamido group at meta position (compound 27: IC50 = 0.39 μM) significantly diverged from para acetamido derivative (compound 26: IC50 = 5.7 μM), where the acetamido function tolerated only at the meta position. A swing in the meta vs para trail was observed with the chloro substituted positional isomers [29 (IC50 = 27.2 μM) vs 30 (IC50 = 0.58 μM)], where meta-Cl containing 29 resulted in a ∼47-fold drastic loss in potency. Although meta vs para trail could not justify the effect of fluoro substituted positional isomers (compounds 32–33), surprisingly, 3,4-difluorobenzyl variant 31 possessed a significant detrimental effect on the PLpro inhibitory potency (IC50 = 29.2 μM). The additional fluoro function might increase the negative charge characteristic of compound 31, thereby tailed off its PLpro inhibitory activity.
Implications of naphthyl derivatives as SARS-CoV-2 PLpro inhibitors
PLpro inhibitors have the potential to be broad spectrum inhibitors due to high sequence similarities of PLpro enzyme in different CoVs. It is interesting to know whether these naphthyl derivatives are effective also in SARS-CoV-2 PLpro target along with SARS-CoV PLpro. We have done both molecular docking and molecular dynamics simulations of three naphthyl derivatives, compounds 23, 27 and 32, against SARS-CoV-2 PLpro target (PDB: 6WUU). The energy minimized geometry of naphthyl derivatives were considered for the docking experiments against SARS-CoV-2 PLpro by the aid of AutoDock Vina [45]. The docking results are depicted as Fig. 6.

Superimposition of docking poses of compounds 23, 27 and 32 (yellow) in PLpro SARS-CoV-2 (PDB: 6WUU, marine blue) in surface representation a and interactions with important amino acids in the binding site b
Figure 6 highlighted the superimposition of docking poses of compounds 23, 27 and 32 in SARS-CoV-2 PLpro (PDB: 6WUU). The docking analysis clearly highlights that naphthyl derivatives can nicely bind with the binding pocket of SARS-CoV-2 PLpro.
MD simulations
After the selection of the lowest energy conformation from the docking output of compounds 23, 27 and 32 (Fig. 7a–c), the stability of the protein structure with those ligand conformations was analysed by MD simulations. The calculated average RMSD of the apo, prt-23, prt-27, and prt-32 are 0.320, 0.315, 0.309, and 0.357 nm, respectively. Data of the average RMSD showed that each of the complex protein structure has almost similar structural deviations like the apo form during dynamics (Fig. 7d). But as depicted in the Fig. 7d, the backbone deviation of protein structure is more stable with the compound 23 in comparison of other compounds. Average RMSF in the residues of the apo, prt-23, prt-27 and prt-32 are calculated to be 0.142, 0.132, 0.133 and 0.143 nm during the dynamics, respectively (Fig. 7e). These revealed that the fluctuations in the residues of protein are more relaxed after binding of compounds 23 and 27 as compared to compound 32 that presented fluctuation similar to apo protein. To analyse the induced changes in the compactness of protein structure after binding of ligand, the Rg data of each complex is compared with that of the apo form (Fig. 7f). The average Rg in the backbone structure of the apo, prt-23, prt-27, and prt-32, are enumerated as 2.338, 2.327, 2.330, and 2.305 nm, respectively, which showed that the compactness of the structure in the apo and complexes in similar during the dynamics. The plotted data (Fig. 7f) depicted that the compactness of the protein backbone is slightly better in the presence of compound 32 in comparison to other compounds and apo protein.

(a–c) Detailed ligand receptor interactions for compounds 23 a, 27 b and 32 c in PLpro SARS-CoV-2 (PDB: 6WUU). (d–f) Molecular dynamics plots are showing RMSD d, RMSF e and Rg f of the backbone-atoms of the apoPLpro SARS-CoV2 and its complexes. In MDS plots, apo, prt-23, prt-27, and prt-32 are represented as red, green, black and blue lines, respectively [here, prt = PLpro]
Hence, comparative MD data analysis delineated that the protein bound with compounds has retained apo-like backbone deviations and compactness along with low fluctuations in the residues throughout the dynamics. Additionally, stability of each ligand conformation in the active site of the PLpro substantiates the docking studies.
The g_MMPBSA tool of the GROMACS is also used to determine the affinity of the compounds with protein as well as the free energy terms responsible for the affinity (Table 4).
The binding energy analysis showed that the compound 32 has more affinity towards protein during the dynamics in comparison with other compounds. Throughout the simulation, both of the electrostatic and van der Waals energy terms contributed majorly for the affinity between the compound and protein in the complexes.
This above analysis proves that naphthyl derivatives have potential to be inhibitor against SARS-CoV-2 PLpro enzyme. All the structural attributes identified by our different modelling approaches may be valid for SARS-CoV-2 PLpro enzyme also. Therefore, it can be emphasized that the naphthyl derivatives have potential to use as a seed for ligand design as well as optimization against SARS-CoV-2 PLpro enzyme by taking different modelling insights performed in this study.
Conclusion
Human coronavirus infections had almost been forgotten, and it was not challenged until novel coronavirus outbreak in December 2019. As per the recent World Health Organization reports, the novel corona virus may never be wiped out completely from the world. There are various strategies including drug repurposing, vaccine and immunity approaches, etc., employed to quickly react in the situation. However, scientific community should also give serious thought to start finding inhibitors against the different targets of the virus through rational drug discovery approaches. In this connection, the inhibitors already designed against different targets of previous human coronavirus infections will be a great starting point for further optimization. In spite of few drugs being assessed clinically against COVID-19, there remains thirst for discovering new molecules with increased efficacy as well as safety. Our research unit previously demonstrated the quantitative structure–activity relationship studies on SARS-CoV protease inhibitors [31,32,33,34].
Here, we endorsed rational drug design efforts through computational drug discovery approaches including machine learning and molecular docking studies. The different molecular modelling techniques such as structural and physico-chemical interpretation (SPCI) analysis and Monte Carlo optimization-based QSAR study collectively deliver some crucial structural information modulating SARS-CoV PLpro inhibitory activities. By considering all these QSAR models, molecular docking and MD simulation studies, it can be concluded that:
(1) Presence of 1-naphthyl head affects the activity, since it modulates the interactions at the active site residues (Fig. 8). This conclusion is based on SPCI analysis, Monte Carlo optimization-based QSAR and docking studies.

The requisite structural features has to be considered to design new molecules with improved activity
(2) The presence of a piperidine moiety is important for binding interaction, since it plays a significant role in interaction with active site residues as suggested by the molecular docking study (Fig. 8).
(3) The stereo-chemical pattern of the methyl substituent at R3 position is a critical factor to modulate PLpro binding affinity (Fig. 8). This can be understood by the docking study. Similarly, (R)-1-naphthylethylamide function is important since it interacts with amino acid residues Tyr-265 and Tyr-269 aromatic rings and with side chain of amino acid residues Pro-248 and Pro-249 and consequently, forming hydrophobic interactions.
(4) The unsubstituted R4 position is more favourable for PLpro inhibition.
(5) The amino function at the linker area is important to form hydrogen bond interaction with PLpro active site residue as evidence by the molecular docking study.
(6) Presence of p-NH2 alone at the R2 position might hamper the binding with the PLpro enzyme.
(7) 5-Methylamine and halogen substituent at the R2 position are beneficial for the PLpro inhibitory activity.
(8) Methoxy substation at the R2 position is unfavourable for biological activity towards PLpro enzyme. This conclusion is based on SPCI analysis and Monte Carlo optimization-based QSAR studies (Fig. 8).
Significantly, the above requisite structural features (Fig. 8) enlighten the perspective of medicinal chemists to develop potent PLpro inhibitors in the future. Research funding agencies and industries should consider in vitro and in vivo studies of the investigated naphthyl derivatives as a seed which sustain significant hope against SARS-CoV-2.
References
Benvenuto D, Giovanetti M, Salemi M, Prosperi M, De Flora C, Junior Alcantara LC, Angeletti S, Ciccozzi M (2020) The global spread of 2019-nCoV: a molecular evolutionary analysis. Pathog Glob Health 114:64–67. https://doi.org/10.1080/20477724.2020.1725339
Ojha PK, Kar S, Krishna JG, Roy K, Leszczynski J (2020) Therapeutics for COVID-19: from computation to practices—where we are, where we are heading to. Mol Divers. https://doi.org/10.1007/s11030-020-10134-x
Amin SA, Jha T (2020) Fight against novel coronavirus: a perspective of medicinal chemists. Eur J Med Chem 201:112559. https://doi.org/10.1016/j.ejmech.2020.112559
https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (as accessed on 26th Nov 2020)
https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (as accessed on 26th Nov 2020)
Ghosh AK, Brindisi M, Shahabi D, Chapman ME, Mesecar AD (2020) Drug development and medicinal chemistry efforts toward SARS-Coronavirus and Covid-19 therapeutics. Chem Med Chem 15:907–932. https://doi.org/10.1002/cmdc.202000223
De P, Bhayye S, Kumar V, Roy K (2020) In silico modeling for quick prediction of inhibitory activity against 3CLpro enzyme in SARS CoV diseases. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1821779
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412. https://doi.org/10.1126/science.abb3405
Báez-Santos YM, John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38. https://doi.org/10.1016/j.antiviral.2014.12.015
Lin MH, Moses DC, Hsieh CH, Cheng SC, Chen YH, Sun CY, Chou CY (2018) Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antiviral Res 150:155–163. https://doi.org/10.1016/j.antiviral.2017.12.015
Park JY, Kim JH, Kim YM, Jeong HJ, Kim DW, Park KH, Kwon HJ, Park SJ, Lee WS, Ryu YB (2012) Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg Med Chem 20:5928–5935. https://doi.org/10.1016/j.bmc.2012.07.038
Chou CY, Chien CH, Han YS, Prebanda MT, Hsieh HP, Turk B, Chang GG, Chen X (2008) Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol 75:1601–1609. https://doi.org/10.1016/j.bcp.2008.01.005
Cheng KW, Cheng SC, Chen WY, Lin MH, Chuang SJ, Cheng IH, Sun CY, Chou CY (2015) Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antiviral Res 115:9–16. https://doi.org/10.1016/j.antiviral.2014.12.011
Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD (2006) Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA 103:5717–5722. https://doi.org/10.1073/pnas.0510851103
Harcourt BH, Jukneliene D, Kanjanahaluethai A, Bechill J, Severson KM, Smith CM, Rota PA, Baker SC (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol 78:13600–13612. https://doi.org/10.1128/JVI.78.24.13600-13612.2004
Chen X, Chou CY, Chang GG (2009) Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir Chem Chemother 2009:151–156. https://doi.org/10.1177/095632020901900402
Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK, Li K (2010) Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 84:4619–4629. https://doi.org/10.1128/JVI.02406-09
Yang X, Chen X, Bian G, Tu J, Xing Y, Wang Y, Chen Z (2014) Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J Gen Virol 95:614–626. https://doi.org/10.1099/vir.0.059014-0
Niemeyer D, Mösbauer K, Klein EM, Sieberg A, Mettelman RC, Mielech AM, Dijkman R, Baker SC, Drosten C, Müller MA (2018) The papain-like protease determines a virulence trait that varies among members of the SARS-coronavirus species. PLoS Pathog 14:e1007296. https://doi.org/10.1371/journal.ppat.1007296
Park JY, Jeong HJ, Kim JH, Kim YM, Park SJ, Kim D, Park KH, Lee WS, Ryu YB (2012) Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biol Pharm Bull 35:2036–2042. https://doi.org/10.1248/bpb.b12-00623
Cho JK, Curtis-Long MJ, Lee KH, Kim DW, Ryu HW, Yuk HJ, Park KH (2013) Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg Med Chem 21:3051–3057. https://doi.org/10.1016/j.bmc.2013.03.027
Lee H, Lei H, Santarsiero BD, Gatuz JL, Cao S, Rice AJ, Patel K, Szypulinski MZ, Ojeda I, Ghosh AK, Johnson ME (2015) Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem Biol 10:1456–1465. https://doi.org/10.1021/cb500917m
Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC (2008) A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA 105:16119–16124. https://doi.org/10.1073/pnas.0805240105
Báez-Santos YM, Barraza SJ, Wilson MW, Agius MP, Mielech AM, Davis NM, Baker SC, Larsen SD, Mesecar AD (2014) X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J Med Chem 57:2393–2412. https://doi.org/10.1021/jm401712t
Ghosh AK, Takayama J, Rao KV, Ratia K, Chaudhuri R, Mulhearn DC, Lee H, Nichols DB, Baliji S, Baker SC, Johnson ME (2010) Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein−ligand X-ray structure and biological evaluation. J Med Chem 53:4968–4979. https://doi.org/10.1021/jm1004489
Ghosh AK, Takayama J, Aubin Y, Ratia K, Chaudhuri R, Baez Y, Sleeman K, Coughlin M, Nichols DB, Mulhearn DC, Prabhakar BS (2009) Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome−coronavirus papain-like protease. J Med Chem 52:5228–5240. https://doi.org/10.1021/jm900611t
Wilson and Gisvolds (2004) Textbook of organic medicinal and pharmaceutical chemistry. Lippincott, Williams and Wilkins, Philadelphia
https://www.drugbank.ca/drugs/DB00735(as accessed on 5th May 2020)
https://www.drugbank.ca/drugs/DB00607(as accessed on 5th May 2020)
Freitas BT, Durie IA, Murray J, Longo JE, Miller HC, Crich D, Hogan RJ, Tripp RA, Pegan SD (2020) Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect Dis 6:2099–2109. https://doi.org/10.1021/acsinfecdis.0c00168
Amin SA, Banerjee S, Adhikari N, Jha T (2020) First structure-activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: an endeavour on COVID-19 drug discovery. Mol Diver in press
Amin SA, Ghosh K, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: monte carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1780946
Amin SA, Banerjee S, Ghosh K, Gayen S, Jha T (2020) Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg Med Chem in press. https://doi.org/10.1016/j.bmc.2020.115860
Ghosh K, Amin SA, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J Mol Struct 1224:129026. https://doi.org/10.1016/j.molstruc.2020.129026
Polishchuk P, Tinkov O, Khristova T, Ognichenko L, Kosinskaya A, VarnekKuz’min AV (2016) Structural and physico-chemical interpretation (SPCI) of QSAR models and its comparison with matched molecular pair analysis. J Chem Inf Model 56:1455–1469. https://doi.org/10.1021/acs.jcim.6b00371
Polishchuk P (2017) Interpretation of quantitative structure–activity relationship models: past, present, and future. J Chem Inf Model 57:2618–2639. https://doi.org/10.1021/acs.jcim.7b00274
Baidya AT, Ghosh K, Amin SA, Adhikari N, Nirmal J, Jha T, Gayen S (2020) In silico modelling, identification of crucial molecular fingerprints, and prediction of new possible substrates of human organic cationic transporters 1 and 2. New J Chem 44:4129–4143. https://doi.org/10.1039/C9NJ05825G
Toropov AA, Toropova AP, Lombardo A, Roncaglioni A, Benfenati E, Gini GI (2011) CORAL: Building up the model for bioconcentration factor and defining it’s applicability domain. Eur J Med Chem 46:1400–1403. https://doi.org/10.1016/j.ejmech.2011.01.018
García J, Duchowicz PR, Rozas MF, Caram JA, Mirífico MV, Fernández FM, Castro EA (2011) A comparative QSAR on 1, 2, 5-thiadiazolidin-3-one 1, 1-dioxide compounds as selective inhibitors of human serine proteinases. J Mol Graph Model 31:10–19. https://doi.org/10.1016/j.jmgm.2011.07.007
Toropov AA, Toropova AP, Benfenati E (2010) SMILES-based optimal descriptors: QSAR modeling of carcinogenicity by balance of correlations with ideal slopes. Eur J Med Chem 45:3581–3587. https://doi.org/10.1016/j.ejmech.2010.05.002
Toropov AA, Toropova AP, Benfenati E (2008) QSPR modeling for enthalpies of formation of organometallic compounds by means of SMILES-based optimal descriptors. Chem Phys Lett 461:343–347. https://doi.org/10.1016/j.cplett.2008.07.027
https://github.com/DrrDom/rspci (as accessed on 31st March 2020)
Toropova AP, Toropov AA, Benfenati E (2015) A quasi-QSPR modelling for the photocatalytic decolourization rate constants and cellular viability (CV%) of nanoparticles by CORAL. SAR QSAR Environ Res 26:29–40. https://doi.org/10.1080/1062936X.2014.984327
Worachartcheewan A, Mandi P, Prachayasittikul V, Toropova AP, Toropov AA, Nantasenamat C (2014) Large-scale QSAR study of aromatase inhibitors using SMILES-based descriptors. Chemometr Intell Lab Syst 138:120–126. https://doi.org/10.1016/j.chemolab.2014.07.017
Trott O, Olson AJ (2010) AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem 31:455–461. https://doi.org/10.1002/jcc.21334
http://mgltools.scripps.edu/ (as accessed on 15th Nov 2020)
DS Visualizer 3.5, Accelrys Software Inc., San Diego, California, USA
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software X 1:19–25. https://doi.org/10.1016/j.softx.2015.06.001
van Gunsteren WF (1996) Biomolecular simulation: the GROMOS96 manual and user guide. Biomos Zürich, Zürich, Switzerland, pp 1–1044
http://prodrg2.dyndns.org/submit.html (as accessed on 15th Nov 2020)
Berendsen HJ, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271. https://doi.org/10.1021/j100308a038
Kumari R, Kumar R, Lynn A, Open Source Drug Discovery Consortium (2014) g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Info Model. 54:1951–1962
Acknowledgment
Sk. Abdul Amin sincerely acknowledges Council of Scientific and Industrial Research (CSIR), New Delhi, India, for awarding the Senior Research Fellowship (SRF) [FILE NO.: 09/096(0967)/2019-EMR-I, Dated: 01-04-2019]. Tarun Jha is also thankful for the financial support from RUSA 2.0 of UGC, New Delhi, India, to Jadavpur University, Kolkata, India. We are very much thankful to the Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India, and Department of Pharmaceutical Sciences, Dr. Harisingh Gour University, India, for providing the research facilities. The authors acknowledge the Centre for Modelling Simulation & Design (CMSD), University of Hyderabad, Hyderabad for computational resources for MD simulation studies.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary file 2 (MP4 685 kb)
Supplementary file 3 (MP4 747 kb)
Supplementary file 4 (MP4 706 kb)
Supplementary file 5 (MP4 596 kb)
Rights and permissions
About this article

Cite this article
Amin, S.A., Ghosh, K., Singh, S. et al. Exploring naphthyl derivatives as SARS-CoV papain-like protease (PLpro) inhibitors and its implications in COVID-19 drug discovery. Mol Divers 26, 215–228 (2022). https://doi.org/10.1007/s11030-021-10198-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10198-3