
Abstract
Short synthetic peptide molecules which bind to a specific target protein with a high affinity to exert its function are known as peptide aptamers. The high specificity of aptamers with small-molecule targets (metal ions, dyes and theophylline; ATP) is within 1 pM and 1 μM range, whereas with the proteins (thrombin, CD4 and antibodies) it is in the nanomolar range (which is equivalent to monoclonal antibodies). The recently identified coronavirus (SARS-CoV-2) genome encodes for various proteins, such as envelope, membrane, nucleocapsid, and spike protein. Among these, the protein necessary for the virus to enter inside the host cell is spike protein. The work focuses on designing peptide aptamer targeting the spike receptor-binding domain of SARS-CoV-2. The peptide aptamer has been designed by using bacterial Thioredoxin A as the scaffold protein and an 18-residue-long peptide. The tertiary structure of the peptide aptamer is modeled and docked to spike receptor-binding domain of SARS CoV2. Molecular dynamic simulation has been done to check the stability of the aptamer and receptor-binding domain complex. It was observed that the aptamer binds to spike receptor-binding domain of SARS-CoV-2 in a similar pattern as that of ACE2. The aptamer–receptor-binding domain complex was found to be stable in a 100 ns molecular dynamic simulation. The aptamer is also predicted to be non-antigenic, non-allergenic, non-hemolytic, non-inflammatory, water-soluble with high affinity toward ACE2 than serum albumin. Thus, peptide aptamer can be a novel approach for the therapeutic treatment for SARS-CoV-2.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Short synthetic peptide or oligomolecules that bind to a specific target protein with the high affinity that differs from natural riboswitches to exert its function were aptamers. Nucleotide aptamers are 15–70 nucleotides, whereas peptide aptamers are 5–20 residues long [1]. The high specificity of aptamers with the target (metal ions, dyes, and theophylline; ATP) was brought by interactions in three-dimensional folding through hydrogen bonding, hydrophobic, columbic and van der Waals forces within 1 pM and 1 μM range, whereas with the proteins (thrombin, CD4 and antibodies) it is in the nanomolar range (which is equivalent to monoclonal antibodies) [2]. Their working principle is similar to the lock-and-key hypothesis for the detection of specific molecules, such as antibodies. Oligonucleotide aptamers are chemically synthesized using systematic evolution of ligands by exponential enrichment (SELEX), which are relatively easy with higher affinity and have better specificity inexpensive and lower immunogenicity and nonspecific cross-reactivity to their targets [3,4,5]. Pegaptanib is a chemically synthesized nucleotide aptamer for the treatment for age-related macular degeneration (AMD). Peptide aptamers reveal many advantages, such as detection of toxic compounds or even non-immunogenic targets [6]; they are heat stable, pH stable and more resistant to organic solvents [7,8,9]. Aptamers can be linked to ribozymes termed as aptazymes to knock out gene expression [10, 11]. They were chemically coupled to fluorophores and radioisotope reporters at 5′ or 3′ end on the DNA backbone for diagnosis and therapy [12, 13]. The disadvantage is a short half-life time, and due to their small size, they promote renal clearance and nuclease degradation [14]. Applications include biotechnology [15], toxicology [16], short-term test for cocaine [15], diagnostics [16], drugs, ophthalmology [17], oncology [18] and anticoagulant therapy [19,20,21]. The functioning of NS3 helicase domain of the hepatitis C virus (HCV) was inhibited by RNA aptamers [22, 23].
The recently identified novel coronavirus (SARS-CoV-2), the seventh coronavirus, known to infect humans is originated from Wuhan (China) in December 2019 [24] which consists of a single-stranded RNA genome with a lipid coat about 100–120 nM [25]. The genome encodes for various proteins, such as encapsulated envelope (E) protein, outer membrane (M) protein, inner nucleocapsid (N) protein and projected spike (S) protein. Among these, the protein necessary for the virus to enter inside the host cell is the spike protein (S), a homotrimeric 1273 amino acids [26]. Spike protein-dependent entry inside the cell involves the attachment of the virus to the host; alteration of protein conformation; proteolysis; release of S2 subunit of the spike protein; and endocytosis [27, 28]. Using co-immunoprecipitation technique, the role of angiotensin-converting enzyme 2 (ACE2) as a receptor in SARS-CoV infections was studied [29, 30]. Spike protein binds to ACE2 receptor of host surface through S1 subunit of receptor-binding domain (RBD) [31,32,33] which exposes cleavage site on S2 subunit that is acted on by host cell proteases to initiate the process of cellular entry [26, 34]. The RBD binds to the carboxypeptidase domain of ACE2 and moves like a hinge between up and down to expose or hide [35], and cleavage occurs first through priming then activation [27, 28]. The cleavage site between S1 and S2 of SARS-CoV-2 occurs at Arg-Arg-Ala-Arg residue between 682 and 685 amino acid regions, which are essential to activate the protein for membrane fusion. Viral spreading also occurs through receptor-dependent cellular fusion to the host [36]. ACE2 is a type 1 transmembrane protein with a zinc-binding domain and expressed in the lung, heart, kidney, testis and gastrointestinal tract [37, 38]. The lung ACE2 [39] appears to have distinct physiological functions from ACE [37, 40].
Since ACE2 and RBD of S-protein interaction are important for SARS-CoV-2 entry into the host cell, peptide aptamer mimicking ACE2 can be used to inhibit the interaction between ACE2 and RBD of S-protein. For designing a peptide aptamer, there is a need for scaffold protein, which does not affect solubility, folding and aggregation properties, and is highly preferred. For conformationally constrained peptides, bacterial Thioredoxin A (TrxA) was designed [41]. Thioredoxin A is 12 kDa oxidoreductase enzyme involved in thiol/disulfide equilibrium in E. coli. Its active site has Cys-Gly-Pro-Cys stretch that accommodates preferably long peptide insertions with the loss of enzymatic function [42]. It consists of a β-strand surrounded by α-helices with an active site on the surface [43].
In this paper, we propose to study the interaction between peptide aptamer and RBD of the S-protein of CoV2 in silico. The peptide aptamer can be injected directly into the bloodstream of the host or can be administered through the nasal cavity. Interaction of the peptide aptamer with S-protein receptor-binding domain will inhibit the interaction of S-protein with ACE2. This will in turn inhibit viral entry to the host cell. Thus, peptide aptamer can be a novel therapeutic approach in SARS-CoV-2 infection.
Materials and methods
Designing of the peptide aptamer
Peptide aptamer consists of two parts: a scaffold protein and a 5–20 residue peptide sequence binding to a particular target. Bacterial Thioredoxin A (UniProt ID P0AA25 and PDB ID 1KEB) was selected as a scaffold protein. The active site of Thioredoxin A consisting of the sequence CGPC was the point of insertion of the peptide aptamer. The SARS-CoV-2 spike protein binds to ACE2 through the receptor-binding domain. The region of ACE2 involved in binding to the SARS-CoV-2 receptor-binding domain was selected as an aptamer. The aptamer sequence is 18 residues long consisting of the sequence N-AKTFLDKFNHEAEDLFYQ-C and inserted between CGPC motifs of Thioredoxin A.
Physiochemical properties and solubility prediction
Physiochemical properties such as pI, half-life, amino acid arrangement, instability and aliphatic index are the molecular weight of the construct and are predicted using ProtParam web server (https://web.expasy.org/protparam/). Grand average of hydropathicity (GRAVY) is calculated in GRAVY Calculator web server (http://www.gravy-calculator.de/).
Secondary structure prediction
PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) and RaptorX Property [44] are used for the secondary structure prediction of the peptide aptamer. PSIPRED involves incorporating two feed-forward neural networks in which analysis output was obtained from PSI-BLAST (position-specific iterated-BLAST). A template-free approach using the RaptorX Property web server was further used to predict the secondary structure properties of the peptide aptamer. The server is enhanced by a machine learning model called DeepCNF (deep convolutional neural fields) to simultaneously predict secondary structure, solvent accessibility and disorder regions.
Three-dimensional structure prediction
Tertiary structure modeling of peptide aptamer was carried out using I-TASER (Iterative Threading ASSEmbly Refinement) server [45,46,47] that is an integrated platform for automated protein structure and function prediction. I-TASER generates three-dimensional (3D) fragments of atomic models from the primary sequence by multiple threading alignments and then reassembles into full-length models by replica-exchange Monte Carlo simulations.
Refinement of the tertiary structure
The three-dimensional model obtained for the peptide aptamer was refined in the GalaxyRefine server [48] which can improve the quality of both global and local tertiary structures that start with rebuilding side chains, repacking and then performing molecular dynamic simulation for overall structure relaxation.
Three-dimensional structure validation
Three-dimensional structure validation detects errors in predicted three-dimensional models performed using SAVES v5.0 server (https://servicesn.mbi.ucla.edu/SAVES/). SAVES v5.0 server gives ERRAT score to analyze non-bonded atom–atom interactions compared to reliable high-resolution crystallography structures. A Ramachandran plot was obtained to visualize dihedral angles psi (ψ) and phi (ϕ) of amino acid. Verifying three-dimensional score obtained from this server determines the compatibility of an atomic model (3D) with its primary sequence (1D).
Molecular docking for peptide aptamer and SARS-CoV-2 S-protein RBD
Molecular docking for peptide aptamer and SARS-CoV-2 S-protein RBD (PDB ID 6M0J) is performed using ClusPro 2.0 web server to evaluate the interaction between the two proteins [49]. ClusPro 2.0 web server generates billions of complexes from which the clusters with the best electrostatic and desolvation energies are filtered, and the docked complex is visualized in the Biovia Discovery Studio R2 software package. The binding free energy of the docked complex was calculated in PRODIGY (protein-binding energy prediction) web server (https://bianca.science.uu.nl/prodigy/) which is a collection of web services focused on the prediction of binding affinity in biological complexes as well as the identification of biological interfaces from crystallographic one.
Molecular dynamic simulation
The aptamer–RBD complex then underwent 100 ns molecular dynamic simulation using GROMACS 4.6.5 software packages [50]: CHARMM forcefield and SPC/E water model. The simulation system was neutralized by adding a suitable number of Na+/Cl− ions. The solvated system was energy minimized in 1000 steps using steepest descent method iterations. After setting the temperature at 300 K and pressure at 1 bar, production simulation was run for 100 ns to evaluate dynamics of peptide aptamer, RBD of S-protein of SARS-CoV-2 as well as the aptamer–RBD complex. RMSD profile of peptide aptamer, RBD of S-protein of SARS-CoV-2 and aptamer–RBD complex and hydrogen-bonding profile of aptamer–RBD complex were studied.
MM-PBSA method of calculating binding energy
Binding free energy calculation was done in g_mmpbsa tool [51] for the last 10 ns of the production run. Results are calculated as average with standard deviation. The peptide aptamer in the aptamer–RBD complex is considered as a ligand.
Biocompatibility prediction of the designed peptide aptamer
To predict the biocompatibility of the peptide aptamer, solubility prediction, hemolytic probability, anti-inflammatory potential and plasma protein binding were studied. Solubility prediction was performed in Protein-Sol server [52]. The average solubility of the experimental dataset used in the server is 0.45. Protein with a value higher than 0.45 will be considered as a soluble protein. Since the aptamer will be circulated via the bloodstream, there will be a frequent collision between plasma proteins and the aptamer. To study the impact of these collisions, a docking study of human serum albumin (the most abundant plasma protein) and peptide aptamer was performed in ClusPro 2.0 server [49]. The structure of human serum albumin (HSA) was retrieved from RCSB PDB with ID 1AO6. Red blood cells constitute about half of the blood volume. Hemoglobin is a well-known protein localized in the red blood cells. Hemolysis is a term used to describe the destruction of red blood cells, and agents that cause hemolysis are called hemolytic agents. Since the aptamer will be released into the bloodstream, the hemolytic probability of the aptamer has been predicted in HemoPI server [53]. The anti-inflammatory potential of the peptide aptamer was predicted using the AIPred server [54].
In silico cloning optimization of the peptide aptamer
The designed peptide aptamer is expressed in a suitable prokaryotic host. Codon optimization is necessary due to codon biasness for the expression of the protein in a prokaryotic host and performed using Java Codon Adaptation Tool (JCat) server (http://www.prodoric.de/JCat) for expression in E. coli K12 host. Transcription termination, ribosome-binding site, and restriction enzyme cleavage sites are avoided during the process. Codon adaptation index (CAI) and percentage GC content were also received as output along with the optimized nucleotide sequence. The optimized nucleotide sequence of the peptide aptamer was cloned into the E. coli pET-28a (+) vector using the SnapGene tool, and Nde I and Bgl II restriction sites were introduced to the N- and C-terminals of the sequence, respectively.
Results
Peptide aptamer and its physiochemical properties
The peptide aptamer was designed using bacterial Thioredoxin A (Fig. 1a, b), and the region of ACE2 binds to SARS-CoV-2 RBD. The designed peptide aptamer consisted of 127 amino acids (14.005 kDa) with an isoelectric point (pI) of 4.08. The protein was predicted to be slightly acidic, and half-life was estimated to be 30 h in mammalian reticulocytes in vitro, > 20 h in yeast and > 10 h in E. coli in vivo. An instability index (II) was predicted to be 13.65, indicating that aptamer is stable. The estimated aliphatic index was predicted to be 96.14, indicating that the aptamer is thermotolerant. The predicted GRAVY was − 0.092, indicating that the protein is hydrophilic and interacts with water molecules.

a Primary sequence of the peptide aptamer. b X-ray crystallographic structure of bacterial Thioredoxin A (PDB ID 1KEB), the scaffold protein. c Modeled structure of peptide aptamer. The sequence AKTFLDKFNHEAEDLFYQ was represented in yellow color. Integration of the peptide sequence into the scaffold protein does not change the native structure of the scaffold protein. d Ramachandran plot showing the distribution of residues of the peptide aptamer in different quadrants
Two-dimensional structure prediction
The peptide aptamer is predicted to contain 41% α-helix, 22% β-strand, and 36% random coil. In addition, 40% of the residues were predicted to be solvent-exposed, 27% exposed to medium and 31% buried inside. A total of five residues (3%) were predicted to be disordered domains by the RaptorX Property server. The secondary structure was predicted by PSIPRED as shown in Fig. 2a. The sequence similarity of aptamer and ACE2 is shown in Fig. 2b. Since the scaffold of the aptamer is a bacterial protein Thioredoxin A, the scaffold does not share any sequence similarity with ACE2. The only similar sequence between peptide aptamer and ACE2 is the sequence of ACE2 inserted in the scaffold protein while designing the aptamer.

a Representation of secondary structure features of peptide aptamer. b Sequence similarity between peptide aptamer and ACE2. The sequence alignment has been performed in Clustal Omega web server (https://www.ebi.ac.uk/Tools/msa/clustalo/)
Tertiary structure modeling
Tertiary structure prediction by I-TASSER server predicted five three-dimensional structure models of the peptide aptamer. A ten threading templates were used for the prediction, in which 3dxbD, 1t00A, 6ib1, 5hr3A and 2trxA were the best templates. All the templates showed good alignment as per Z-score values, ranging from 1.01 to 4.73 and calculated C-score ranging from − 0.27 to − 2.54 which is generally between − 5 and 2; the higher the value , the higher the confidence [55]. Thus, the model with the highest C-score was selected for further studies (Fig. 1c). This model had a TM score of 0.68 ± 0.12 with an estimated RMSD of 5.0 ± 2.3 Å, which is the measure of structural similarity between two structures [55]. A value greater than 0.5 indicates a model of correct topology and less than 0.17 indicates random similarity [55].
Tertiary structure refinement
The three-dimensional structure of the peptide aptamer was refined on GalaxyRefine server that yields five models. Based on model quality scores for all refined models, model 1 was found to be the best (Fig. 1c). The model quality scores included high-accuracy global distance test (GDT-HA) score of 0.9803, RMSD score of 0.332, MolProbity score of 2.577,a clash score of 22.6, poor rotamers score of 2.9 and Ramachandran plot score of 94.4%. This model had chosen for further analysis.
Three-dimensional structure validation
The quality of the modeled peptide aptamer structure was validated using Ramachandran plot analysis; it revealed 88.3% residues in the most favored region and 9.9% residues in the additional allowed region. Additionally, 0.9% of residues were predicted in allowed regions and only 0.9% in the disallowed region (Fig. 1d). The quality and potential errors in the tertiary structure were verified by ERRAT. The chosen model after refinement had an overall quality factor of 93.2773% with ERRAT. High-resolution three-dimensional structures generally produce values around 95% or higher and for lower resolutions (2.5–3A), the overall quality factor was 91%. Verifying three-dimensional score determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) which also passed the modeled structure as 100% of the residues have averaged 3D–1D score ≥ 0.2.
Prediction of the antigenicity and allergenicity of the peptide aptamer
The aptamer has a scaffold protein, Thioredoxin A from a bacterial source. Thus, the antigenicity of the peptide aptamer and allergenicity of the peptide aptamer were determined. The antigenicity of the aptamer was predicted to be 0.33 using VaxiJen 2.0 server with a bacteria model at a threshold of 0.4, and thus non-antigenic. The peptide aptamer was predicted to be non-allergenic on the AllerTOP v.2 server.
Docking of peptide aptamer and RBD of S-protein of SARS-CoV-2
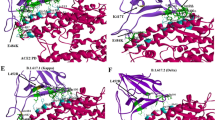
The peptide aptamer docked at the RBD of S-protein of CoV2 binding energy is calculated to be − 11.3 kcal/mol. The binding energy of ACE2 and RBD of S-protein of CoV2 is calculated to be − 11.9 kcal/mol. Thus, the binding energy of aptamer and RBD is close to the binding energy of ACE2 and RBD. Comparison of Fig. 3a, b shows a similar orientation of the designed aptamer and ACE2 on the surface of RBD of S-protein. The comparison of the number of residue contacts of aptamer–RBD complex and ACE2–RBD complex is given in Table 1.

a X-ray crystallographic structure of ACE2–RBD of S-protein of SARS-CoV-2 complex (PDB ID 6M0J). Green color represents RBD, and cyan color represents ACE2. b Docked structure of peptide aptamer–RBD complex. Green color represents RBD, and cyan color represents aptamer. The sequence AKTFLDKFNHEAEDLFYQ was represented in yellow color. c PDBsum analysis of different interactions in ACE2–RBD complex and aptamer–RBD complex (after 100 ns molecular dynamic simulation)
Molecular dynamic simulation
Molecular dynamic simulation is done to check the stability of peptide aptamer and RBD complex. The snapshot of the complex is taken at 0 ns and 100 ns of the simulation showed that the peptide aptamer arranged itself on the RBD of S-protein of CoV2 (Fig. 4a, b). These arrangements took place probably due to the cumulative action of solvent and ions present in the simulation environment. Due to the rearrangement in the binding pattern, residues of aptamer involved in the formation of bonds with RBD have also changed. After 100 ns of simulation, the residues of aptamer involved in bond formation are Lys36, Leu39, Phe42, Asn43, His44, Ala46, Glu47 and Asp48. It was also observed that when the peptide aptamer and the RBD of S-protein of CoV-2 were simulated separately, there were many fluctuations in the RMSD. But when the aptamer–RBD complex was simulated, there was no fluctuation in the RMSD for 100 ns (Fig. 4c). The number of hydrogen bonds was varied throughout the simulation. There was a maximum of 15 hydrogen bonds at some points. During 55–60 ns, the complex experienced the lowest number of intermolecular hydrogen bonds (Fig. 5). The change in residual contact before and after 100 ns simulation is shown in Fig. 6. Thus, the aptamer and RBD complex was stable in an aqueous environment.

a Aptamer–RBD complex at 0 ns. b Aptamer–RBD complex at 100 ns. Green color represents RBD and cyan color represents aptamer. The sequence AKTFLDKFNHEAEDLFYQ was represented in yellow color. c RMSD profile of aptamer, RBD of S-protein of SARS-CoV-2 and aptamer–RBD complex. d Conformation of Lys31, His34, Glu35 and Glu37 of ACE2 upon binding to RBD. e Conformation of Lys41, His44, Glu45 and Glu47 of aptamer upon binding to RBD.

Number of intermolecular hydrogen bonds between peptide aptamer and RBD of S-protein during 100 ns molecular dynamic simulation

Residual contact plot between peptide aptamer and RBD of S-protein a at 0 ns and b at 100 ns molecular dynamic simulation
The residues of the peptide aptamer involved in the interaction with RBD after molecular dynamic simulation are Lys41, Phe42, Asn43, His44, Glu45 and Glu47 (Fig. 3c). The residues of the ACE2 involved in the interaction with RBD are Gln24, Thr27, Phe28, Asp30, Lys31, His34, Glu35, Glu37, Asp38, Tyr41 and Glu42 (Fig. 3c). Thus, four of the RBD-interacting residues of ACE2 (Lys31, His34, Glu35 and Glu37) also interacted with RBD when presented as an aptamer. The residues of ACE2 (Lys31, His34, Glu35 and Glu37) have been numbered as Lys41, His44, Glu45 and Glu47 in the peptide aptamer during modeling. These residues, however, have different conformations when in ACE2 and aptamer. In ACE2, His34 and Glu35 are facing toward opposite direction, but, in aptamer, His44 and Glu45 are facing in the same direction. Lys31 and His34 are in close proximity in ACE2 but Lys41 and His44 are a little far away. Due to alpha-helical conformation, His34 and Glu37 are in close proximity in ACE2. However, in aptamer, His44 and Glu47 are far apart.
Binding free energy calculation using MM-PBSA gave an average binding energy of − 173.019 ± 15.612 kcal/mol for the last 10 ns of the simulation. Different energy parameters that contributed to the binding energy were also calculated. The van der Waals energy was found to be − 74.7522 ± 13.846 kcal/mol, electrostatic energy was found to be − 222.668 ± 16.734 kcal/mol, polar solvation energy was found to be 124.402 ± 9.640 kcal/mol and nonpolar solvation energy was found to be − 53.8351 ± 12.116 kcal/mol.
Biocompatibility of the peptide aptamer
Any drug molecule, whether it is small molecules, antibodies or aptamers, circulated through the bloodstream. Water constitutes the majority of the blood plasma. The designed aptamer is predicted to be soluble with a solubility score of 0.69, which is higher than the experimental data set (Fig. 7a). Plasma protein is another constituent of blood plasma, and serum albumin is a major plasma protein. The peptide aptamer binds with the HSA with a binding energy of − 6.6 kcal/mol, which is much higher than the binding energy of aptamer–RBD of S-protein complex (− 11.3 kcal/mol). It can be predicted that the peptide aptamer will have a higher affinity towards the S-protein of SARS-CoV-2 than HSA. Red blood cells constitute more than half of the blood cells. But, these cells are prone to hemolysis when they come in contact with hemolytic agents. The designed aptamer has 0 (probability range 0–1, 1 being highly probable) hemolytic probability, thus classifying it as a non-hemolytic peptide. The designed aptamer is also predicted as an anti-inflammatory with a probability score of 0.4930 (probability range 0–1, 1 being highly probable).

a Solubility prediction of the designed aptamer as compared to an experimental data set. b In silico cloning of the designed aptamer
Codon optimization of the peptide aptamer
The peptide aptamer had to express in a suitable host for in vitro and in vivo studies. E. coli (strain K12) is a suitable host for the expression of peptide aptamer. Thus, for codon optimization in E. coli K12 host, the Java Codon Adaptation Tool (JCat) used and optimized codon sequence length was 581 nucleotides. The CAI was 1.0 which indicates nucleotide sequence contains the most frequently occurring codons [56]. The average GC content of the optimized sequence was 50.91% and a higher value denotes better expression in prokaryotes [57, 58]. The optimized nucleotide sequence was inserted into the pET28a (+) vector using SnapGene software (Fig. 7b).
Discussion
Aptamers had been developed as alternatives to antibodies [1]. Aptamers work in a similar mechanism as that of antibodies. Aptamer gains an advantage over antibodies in ease of production. Antibodies produced by our body in response to a foreign particle are the primary source of defense. This is the basis of vaccination, where a dead or inactive microorganism or a part of microorganism was injected into the body to generate an immune response. Another way of vaccination is the administration of antibodies (for example, snake antivenom immunoglobulins). In this method, the antibodies produced in a different host in response to the foreign particle are purified, stored and then injected when required. The method of vaccination requires both time and money, and there is an added risk of hyperallergic reaction. To overcome the time limit, cost and associated health risks, aptamers had been developed. Nucleic acid aptamers were synthesized chemically and peptide aptamers can be expressed in a bacterial host. The aptamers bind specifically to targets from the picomolar to micromolar range.
The SARS-CoV-2 outbreak in December 2019 has attracted the attention of worldwide researchers. Researchers are trying to study the effect of small molecules (FDA-approved drugs, natural molecules) on SARS-CoV-2 infection. In silico approach helps in the identification of unique peptides from viral proteins, which were used as the means of strategy for SARS-CoV-2 infection [59]. Several proteins encoded by SARS-CoV-2, such as 3-chymotrypsin-like protease (3CLpro), Spike, RNA-dependent RNA polymerase (RdRp) and papain-like protease (PLpro), were targeted for efficient drug design using computational approaches [60]. The early detection of the virus could be achieved by using biosensors, and the usage of nanovaccines efficiently reduces virus; thus, nanomedicine could be one such strategy for an efficient way of reducing virus [61]. Alternatively, there are extensive researches going on in search of a vaccine. A few vaccine candidates are at different stages in the clinical trial phase. Considering the amount of time taken to develop a vaccine candidate, peptide aptamer has been designed and can be used as an alternate therapy. Although some antiviral drugs, such as remdesivir and baricitinib, are being used, the time taken to repurpose those drugs could not be ignored. Thus, in a pandemic situation, aptamers could be useful as it takes very less time to be produced. The scaffold protein used in the aptamer is already validated for its compatibility with host. Although aptamer research is in a very initial stage, aptamers can become an important therapy in several diseases including viral infections.
In this paper, we have focused on designing of peptide aptamer to inhibit the binding of SARS-CoV-2 spike protein to host ACE2 and allowed the genetic material of the virus to enter inside the host. Peptide aptamer was designed by inserting an 18-residue-long amino acid sequence N-AKTFLDKFNHEAEDLFYQ-C into a scaffold protein, that is, bacterial Thioredoxin A. This inserted sequence in the region of ACE2 is involved in the interaction with RDB of S-protein of SARS-CoV-2. The peptide aptamer was modeled using I-TASSER. Secondary structure prediction of the peptide aptamer revealed 41% α-helix, 22% β-strand, and 36% coil in the aptamer. In addition, 40% of the residues were predicted to be solvent-exposed, 27% medium-exposed and 31% buried. The quality of the tertiary structure was assessed by generating a Ramachandran plot, and it was revealed that 88.3% are present residues in the most favored region and 9.9% residues in the additional allowed region. The designed peptide aptamer was predicted to be non-antigenic and non-allergenic.
The peptide aptamer was docked at the RBD of S-protein of CoV2 with a binding energy of − 11.3 kcal/mol, which is close to the binding energy of ACE2 and RBD of S-protein of CoV2 (− 11.9 kcal/mol). The residues of the peptide aptamer involved in the interaction with RBD are Lys41, Phe42, Asn43, His44, Glu45 and Glu47. These residues are also present in ACE2, and they interacted with SARS-CoV-2 S-protein RBD. Molecular dynamic simulation of 100 ns in an aqueous environment revealed a positional rearrangement of peptide aptamer and RBD complex. After 100 ns of simulation, the residues of aptamer involved in bond formation are Lys36, Leu39, Phe42, Asn43, His44, Ala46, Glu47 and Asp48. It was also observed that the aptamer–RBD complex was stable in 100 ns simulation. Hydrogen-bonding pattern of aptamer and RBD of S-protein revealed a formation of maximum of 14 hydrogen bonds. The calculation of energy parameters using MM-PBSA gave average binding energy of − 173.019 ± 15.612 kcal/mol for the last 10 ns of the simulation. The van der Waals energy was found to be − 74.7522 ± 13.846 kcal/mol, electrostatic energy was found to be − 222.668 ± 16.734 kcal/mol, polar solvation energy was found to be 124.402 ± 9.640 kcal/mol and nonpolar solvation energy was − 53.8351 ± 12.116 kcal/mol. The peptide aptamer is also predicted as soluble, non-hemolytic and anti-inflammatory. Molecular docking with HAS showed that the peptide aptamer has a lower affinity for HAS (plasma protein) as compared to RBD of S-protein.
Therefore, the peptide aptamer could be a potential inhibitor of S-protein of SARS-CoV-2 and host cell ACE2 interaction. The scope of the present study is to design a peptide aptamer that can be used as an alternative to the vaccine. The study is not only limited to designing the aptamer but also includes necessary predictions of the biophysical characteristics of the aptamer. The only limitation of the present study is molecular dynamic simulations where only the RBD of S-protein has been used. The peptide aptamer will be cloned into pET28a(+) vector followed by expression and purification. In vitro and in vivo studies with the purified aptamer will be carried out for further validation.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- ACE:
-
Angiotensin-converting enzyme
- AMD:
-
Age-related macular degeneration
- ATP:
-
Adenosine triphosphate
- CD4:
-
Complementary determining region 4
- CHARMM:
-
Chemistry at Harvard macromolecular mechanics
- DeepCNF:
-
Deep convolutional neural fields
- DNA:
-
Deoxyribose nucleic acid
- GRAVY:
-
Grand average of hydropathicity
- GROMACS:
-
GROningen MAchine for Chemical Simulations
- HCV:
-
Hepatitis C virus
- I-TASSER:
-
Iterative threading assembly refinement
- NS3:
-
Non-structural protein 3
- PRODIGY:
-
Protein-binding energy prediction
- PSIPRED:
-
Position-specific iterated blast-based secondary structure prediction
- PSI-BLAST:
-
Position-specific iterated-basic local alignment search tool
- RBD:
-
Receptor-binding domain
- RNA:
-
Ribonucleic acid
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SELEX:
-
Systematic evolution of ligands by exponential enrichment
- siRNA:
-
Small interfering ribonucleic acid
- SPC/E:
-
Extended simple point charge
- TrxA:
-
Thioredoxin A
- MM-PBSA:
-
Molecular mechanics Poisson–Boltzmann surface area
- HSA:
-
Human serum albumin
References
Reverdatto S, Burz DS, Shekhtman A (2015) Peptide aptamers: development and applications. Curr Top Med Chem 15(12):1082–1101. https://doi.org/10.2174/1568026615666150413153143
Kadioglu O et al (2015) Aptamers as a novel tool for diagnostics and therapy. Invest New Drugs 33(2):513–520. https://doi.org/10.1007/s10637-015-0213-y
Mayer G (2009) The chemical biology of aptamers. Angew Chem Int Ed Engl 48(15):2672–2689. https://doi.org/10.1002/anie.200804643
CollettChoEllington JREJAD (2005) Production and processing of aptamer microarrays. Methods 37(1):4–15. https://doi.org/10.1016/j.ymeth.2005.05.009
Hall DA, Ptacek J, Snyder M (2007) Protein microarray technology. Mech Ageing Dev 128(1):161–167. https://doi.org/10.1016/j.mad.2006.11.021
Lee JH et al (2010) Molecular diagnostic and drug delivery agents based on aptamer-nanomaterial conjugates. Adv Drug Deliv Rev 62(6):592–605. https://doi.org/10.1016/j.addr.2010.03.003
Langer R (1998) Drug delivery and targeting. Nature 392(6679 Suppl):5–10
Wilson C, Szostak JW (1998) Isolation of a fluorophore-specific DNA aptamer with weak redox activity. Chem Biol 5(11):609–617. https://doi.org/10.1016/S1074-5521(98)90289-7
Farokhzad OC et al (2006) Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA 103(16):6315–6320. https://doi.org/10.1073/pnas.0601755103
Thompson KM et al (2002) Group I aptazymes as genetic regulatory switches. BMC Biotechnol 2:21. https://doi.org/10.1186/1472-6750-2-21
Isaacs FJ et al (2004) Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol 22(7):841–847. https://doi.org/10.1038/nbt986
Lee JH et al (2007) Site-specific control of distances between gold nanoparticles using phosphorothioate anchors on DNA and a short bifunctional molecular fastener. Angew Chem Int Ed Engl 46(47):9006–9010. https://doi.org/10.1002/anie.200702569
Ulrich H, Wrenger C (2009) Disease-specific biomarker discovery by aptamers. Cytometry A 75(9):727–733. https://doi.org/10.1002/cyto.a.20766
Famulok M, Hartig JS, Mayer G (2007) Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev 107(9):3715–3743. https://doi.org/10.1021/cr0306743
Gold L et al (2012) Advances in human proteomics at high scale with the SOMAscan proteomics platform. N Biotechnol 29(5):543–549. https://doi.org/10.1016/j.nbt.2011.11.016
Ilyas A et al (2012) Electrical detection of cancer biomarker using aptamers with nanogap break-junctions. Nanotechnology 23(27):275502. https://doi.org/10.1088/0957-4484/23/27/275502
Schachat AP (2005) New treatments for age-related macular degeneration. Ophthalmology 112(4):531–532. https://doi.org/10.1016/j.ophtha.2005.01.018
Chu TC et al (2006) Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res 66(12):5989–5992. https://doi.org/10.1158/0008-5472.CAN-05-4583
Gopinath SC (2008) Anti-coagulant aptamers. Thromb Res 122(6):838–847. https://doi.org/10.1016/j.thromres.2007.10.022
Dobrovolsky AB et al (2009) Inhibition of thrombin activity with DNA-aptamers. Bull Exp Biol Med 148(1):33–36. https://doi.org/10.1007/s10517-009-0627-7
Rusconi CP et al (2002) RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419(6902):90–94. https://doi.org/10.1038/nature00963
Hwang B et al (2004) Isolation of specific and high-affinity RNA aptamers against NS3 helicase domain of hepatitis C virus. RNA 10(8):1277–1290. https://doi.org/10.1261/rna.7100904
Nishikawa F et al (2004) In vitro selection of RNA aptamers against the HCV NS3 helicase domain. Oligonucleotides 14(2):114–129. https://doi.org/10.1089/1545457041526335
Cui J, Li F, Shi ZL (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17(3):181–192. https://doi.org/10.1038/s41579-018-0118-9
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292. https://doi.org/10.1016/S0065-3527(06)66005-3
Walls AC et al (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281-292.e6. https://doi.org/10.1016/j.cell.2020.02.058
Belouzard S, Chu VC, Whittaker GR (2009) Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA 106(14):5871–5876. https://doi.org/10.1073/pnas.0809524106
Millet JK, Whittaker GR (2014) Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci USA 111(42):15214–15219. https://doi.org/10.1073/pnas.1407087111
Kuhn JH et al (2004) Angiotensin-converting enzyme 2: a functional receptor for SARS coronavirus. Cell Mol Life Sci 61(21):2738–2743. https://doi.org/10.1007/s00018-004-4242-5
Li W et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454. https://doi.org/10.1038/nature02145
Wang Q et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4):894-904.e9. https://doi.org/10.1016/j.cell.2020.03.045
Shang J et al (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224. https://doi.org/10.1038/s41586-020-2179-y
Wong SK et al (2004) A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem 279(5):3197–3201. https://doi.org/10.1074/jbc.C300520200
Hoffmann M et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Wrapp D et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507
Pillay TS (2020) Gene of the month: the 2019-nCoV/SARS-CoV-2 novel coronavirus spike protein. J Clin Pathol
Donoghue M et al (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87(5):E1-9. https://doi.org/10.1161/01.RES.87.5.e1
Tipnis SR et al (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275(43):33238–33243. https://doi.org/10.1074/jbc.m002615200
Lan J et al (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581(7807):215–220. https://doi.org/10.1038/s41586-020-2180-5
Hamming I et al (2007) The emerging role of ACE2 in physiology and disease. J Pathol 212(1):1–11. https://doi.org/10.1002/path.2162
Colas P et al (1996) Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature 380(6574):548–550. https://doi.org/10.1038/380548a0
LaVallie ER et al (1993) A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (NY) 11(2):187–193. https://doi.org/10.1038/nbt0293-187
Martin JL (1995) Thioredoxin–a fold for all reasons. Structure 3(3):245–250. https://doi.org/10.1016/s0969-2126(01)00154-x
Wang Z et al (2011) Protein 8-class secondary structure prediction using conditional neural fields. Proteomics 11(19):3786–3792. https://doi.org/10.1002/pmic.201100196
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5(4):725–738. https://doi.org/10.1038/nprot.2010.5
Yang J et al (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12(1):7–8. https://doi.org/10.1038/nmeth.3213
Yang J, Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43(W1):W174–W181. https://doi.org/10.1093/nar/gkv342
Heo L, Park H, Seok C (2013) GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41(Web Server issue):W384–W388. https://doi.org/10.1093/nar/gkt458
Kozakov D et al (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12(2):255–278. https://doi.org/10.1038/nprot.2016.169
Van Der Spoel D et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718. https://doi.org/10.1002/jcc.20291
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10(5):449–461. https://doi.org/10.1517/17460441.2015.1032936
Hebditch M et al (2017) Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 33(19):3098–3100. https://doi.org/10.1093/bioinformatics/btx345
Chaudhary K et al (2016) A web server and mobile app for computing hemolytic potency of peptides. Sci Rep 6:22843. https://doi.org/10.1038/srep22843
Manavalan B et al (2018) AIPpred: sequence-based prediction of anti-inflammatory peptides using random forest. Front Pharmacol 9:276. https://doi.org/10.3389/fphar.2018.00276
Shaik NA et al (2019) Essentials of bioinformatics, volume I: understanding bioinformatics: genes to proteins. doi: https://doi.org/10.1007/978-3-030-02634-9
Lee S et al (2010) Relative codon adaptation index, a sensitive measure of codon usage bias. Evol Bioinform Online 6:47–55. https://doi.org/10.4137/ebo.s4608
Du MZ et al (2018) The GC content as a main factor shaping the amino acid usage during bacterial evolution process. Front Microbiol 9:2948. https://doi.org/10.3389/fmicb.2018.02948
Zhou HQ et al (2014) Analysis of the relationship between genomic GC Content and patterns of base usage, codon usage and amino acid usage in prokaryotes: similar GC content adopts similar compositional frequencies regardless of the phylogenetic lineages. PLoS ONE 9(9):e107319. https://doi.org/10.1371/journal.pone.0107319
Orsburn BC et al (2020) In silico approach toward the identification of unique peptides from viral protein infection: application to COVID-19. bioRxiv. https://doi.org/10.1101/2020.03.08.980383
Wu C et al (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10(5):766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Palestino G et al (2020) Can nanotechnology help in the fight against COVID-19? Expert Rev Anti Infect Ther 18(9):849–864. https://doi.org/10.1080/14787210.2020.1776115
Acknowledgements
AD and NSNC acknowledge Council of Scientific and Industrial Research-Senior Research Fellowship and Indian Council of Medical Research-Senior Research Fellowship for financial assistance, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest..
Rights and permissions
About this article

Cite this article
Devi, A., Chaitanya, N.S.N. Designing of peptide aptamer targeting the receptor-binding domain of spike protein of SARS-CoV-2: an in silico study. Mol Divers 26, 157–169 (2022). https://doi.org/10.1007/s11030-020-10171-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-020-10171-6