
Abstract
The progressive, chronic nature of Alzheimer's disease (AD), a form of dementia, defaces the adulthood of elderly individuals. The pathogenesis of the condition is primarily unascertained, turning the treatment efficacy more arduous. Therefore, understanding the genetic etiology of AD is essential to identifying targeted therapeutics. This study aimed to use machine-learning techniques of expressed genes in patients with AD to identify potential biomarkers that can be used for future therapy. The dataset is accessed from the Gene Expression Omnibus (GEO) database (Accession Number: GSE36980). The subgroups (AD blood samples from frontal, hippocampal, and temporal regions) are individually investigated against non-AD models. Prioritized gene cluster analyses are conducted with the STRING database. The candidate gene biomarkers were trained with various supervised machine-learning (ML) classification algorithms. The interpretation of the model prediction is perpetrated with explainable artificial intelligence (AI) techniques. This experiment revealed 34, 60, and 28 genes as target biomarkers of AD mapped from the frontal, hippocampal, and temporal regions. It is identified ORAI2 as a shared biomarker in all three areas strongly associated with AD's progression. The pathway analysis showed that STIM1 and TRPC3 are strongly associated with ORAI2. We found three hub genes, TPI1, STIM1, and TRPC3, in the network of the ORAI2 gene that might be involved in the molecular pathogenesis of AD. Naive Bayes classified the samples of different groups by fivefold cross-validation with 100% accuracy. AI and ML are promising tools in identifying disease-associated genes that will advance the field of targeted therapeutics against genetic diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that causes brain atrophy and eventually destroys brain cells (Al-Thani et al. 2021). The progression of AD involves cognitive impairments such as memory loss, delusion, disorientation, and confusion. The shrinkage of blood vessels and muscles, inflammation, mitochondrial dysfunction, and production of free radicals are a few reasons that trigger AD (Chethana et al. 2022). AD seems to have a vital genetic component, emphasizing the potential of developing targeted novel therapies to treat AD. According to the National Institute on Aging (NIA), AD is directly linked to Apolipoprotein E (APOE) gene, which triggers AD by disrupting the blood–brain barrier (BBB) integrity (Koutsodendris et al. 2022). However, this gene is not the primary cause of all AD cases. The unknown gene–gene/environment interactions complicate understanding the direct cause of AD. According to age, Alzheimer's is primarily divided into two categories, early-onset and late-onset, where no specific gene is found to impact the disease progression. Recent studies identified a significant association between mutated APP, CD33, and BIN1 genes and the development of AD and neurodegenerative comorbidities (Bhattacharyya et al. 2022).
According to the special report from Alzheimer's Association, 2022, cognitive assessments with the availability of potential blood-based gene biomarkers to aid in early detection and better AD diagnosis. Early AD diagnosis significantly reduces the effect and improves treatment outcomes quickly. This fact exemplifies the potential of gene-based treatment methods that often increases treatment efficacy compared to traditional methods. Several studies have identified several potential biomarkers for AD in recent years. The whole genome sequencing technologies were used to investigate the relationship between cognition-related traits and 174 polymorphisms located on CD36. Six genetically linked variants in the CD36 gene were found to significantly delay the onset of AD (Sery et al. 2022). A ligand library containing 60 natural compounds retrieved from the literature and 25 synthetic compounds from DrugBank is screened for validation against the AD markers. Molecular docking tests identified 11-oxo-tigogenin as the most significant ligand molecule with a binding affinity of -11.1 kcal/mole forming three hydrogen bonds with Arg176, Trp124, and Ile174 (Kushwaha et al. 2021).
The extensive growth in bioinformatics opened up many possibilities for new dimensions of clinical applications (Doss and Zayed 2017; Zaki et al. 2017; Ebrahimi et al. 2018; Thirumal Kumar et al. 2018); Sekaran K et al. 2021). In digital format, multitudinous medical data provide precise insights into any disease. Machine learning (ML) propounds advanced algorithms to solve complex problems in critical domains. In healthcare systems, ML models are widely used to perform clinical diagnosis, biomarker identification, tumor identification, and drug target discovery for various diseases. For instance, since the outbreak of the COVID-19 pandemic, many in-silico studies have been conducted to find novel therapeutics as it takes less time and physical compounds than wet-lab experiments (Bagabir et al. 2022; Yang et al. 2022). The machine learning method Artificial Neural Network (ANN) has been tested to identify the biomarker pattern of Alzheimer's disease. This method predicted gene interaction using a continuous stepwise algorithm and identified fifty potentially influential AD genes in the hippocampus region (Zafeiris et al. 2018). In order to detect capable genes from microarray data and classify AD tissues into different classes, researchers propose a wrapper-based feature selection technique that combines the genetic algorithm with support vector machines (SVM) (Scheubert et al. 2012). This method identifies the twenty most promising candidate markers with three common genes.
Although machine learning (ML) techniques help identify potential disease-associated biomarkers, they require highly developed computational abilities to evaluate their potentiality. Additionally, increased performance is frequently achieved through increased model complexity, transforming such systems into "black box" methods and resulting in uncertainty about how they function and, ultimately, how they make decisions (Linardatos et al. 2020). It is challenging to rely on models whose conclusions cannot be clearly understood. Specific to the medical domain, the AI based clinical decisions about treatment options for a particular condition strongly impact individuals’ well-being. The field of explainable Artificial Intelligence (XAI), which focuses on understanding and interpreting AI systems' behavior, has resurfaced due to the demand for reliable, equitable, robust, high-performing models for real-world applications (Gunning 2019). XAI was used to identify squamous cell carcinoma biomarkers using ML models trained on binary classification datasets containing expression data from healthy and cancer skin samples. Following successful incorporation, 23 significant genes associated with skin cancer progression were discovered, which might serve as diagnostic and prognostic biomarkers. XAI techniques demonstrated that the model output was interpretable by establishing a relationship between the model output and the relevant genes. The explainable clustering and classification approach was used to find and interpret age-based differences in brain tumor diseases (Meena and Hasija 2022). The increased use of ML-based XAI approaches in medical diagnosis has sparked the interest of many future researchers seeking to implement these effective methods in complex diseases.
There is a need to use these advanced computational methods to provide healthcare with valuable biomarkers for early AD detection. Such markers can also shed light on the complex gene network of Alzheimer's pathogenicity. This paper proposes a bioinformatics investigation to identify AD's gene biomarkers from the blood-based gene expression data. We attempt to use gene expression analysis to reduce the dimension of the data statistically and simplify the candidate selection process. The genetic algorithm scrutinizes the DEGs to remove irrelevant features from the subset. The performance and significance of machine learning algorithms trained with extracted biomarker genes will be assessed. The trained models will be interpreted using XAI techniques. Pathway modeling, functional and pathway enrichment, and protein interactions will be performed on the strongly associated genes of identified AD biomarkers.
Materials and methods
Data analysis pipeline
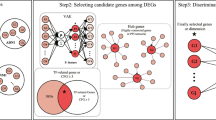
This study aimed to investigate DEGs in patients with AD and identify candidate biomarkers through statistical and machine-learning techniques that might be relevant for treating AD. RNA samples from the blood tissue were collected from three brain regions frontal, temporal and hippocampal. The experimental design of the proposed study includes the following subsections—DEG selection criteria discuss the statistical gene selection procedure, Genetic Algorithm identifies candidate biomarkers of AD through the synthesized natural evolutionary process. The Cytoscape tool performs the gene co-expression analysis. ML algorithms are trained and validated with the gene subset under many steps involved in the task. Explainable artificial intelligence techniques delineate the interpretation of the trained models. Figure 1 visualizes the order of phases involved in the proposed system. This pipeline is implemented in Python (Anaconda Distribution) with supportive ML libraries and XAI modules. The source code of the article is available at https://github.com/karthiksekaran/alzheimer-biomarker-study-XAI.

Data analysis pipeline used in this study. The gene expression data analysis (phase I) and AD biomarker selection using genetic algorithms (phase II). Machine learning, model training, and results interpretation (phase)
Gene expression data collection and analysis
The NCBI-GEO database (http://www.ncbi.nlm.nih.gov/geo/) was used to obtain gene expression data. Hokama et al. (2014) experimental data with accession number GSE36980 was used in this study to train and evaluate the ML models. This data set contains array-based expression profiling data for three different brain regions: frontal (33 samples), hippocampal (18 samples), and temporal (29 samples). This experimental study looked for genes that differed between AD and healthy individuals (Case and Control based study). The credible way to select useful biomarkers in gene expression profiling is by conducting differential gene expression analysis. GEO2R was used to compare two groups of gene expression data, healthy and AD cases, across three different brain regions samples. GEO2R is an interactive web platform supported by NCBI to compare two or more groups of samples available in the GEO series to find DEG from the expression data (Barrett et al. 2012). The DEGs are often expressed in an irregular pattern between two experimental conditions when they are statistically significant. The adjustment is made to the p-value through the Benjamini and Hochberg false discovery rate method. Log transformation is applied, and the typical values are replaced with logarithmic values. This technique normalizes the dataset by addressing skewness trouble. We identified the top 250 DEGs with GEO2R, irrespective of the p-value scale, to avoid rejecting the informative AD biomarkers. Furthermore, to reduce the data used for machine learning model training and to increase diversity.
Biomarker selection using a genetic algorithm
The intrinsic impediment in gene-based computer modeling is finding the most prominent biomarkers from inordinate dimensions of genome information. The genetic algorithm analysis method is widely used to solve search-related optimization problems by performing biological operations such as selection, crossover, and mutation (John 1992). In this study, the genetic algorithm method was used to reduce the high dimensionality of the gene expression data to obtain the optimal subset of features for ML model training. Weka 3.8.2, a java based machine learning software, is used to perform feature selection from the attribute search options. The GA algorithm identified 34, 60, and 28 from frontal, hippocampal, and temporal regions. Based on the observation, the ORAI2 gene was found in all three regions, whereas the TPI1 gene is present only in common between frontal and hippocampal regions. Further experiments on the identified novel AD markers disclosed many associated genes (Table 1). The hypothesis of GA was demonstrated with few requirements, a genetic representation of the solution and a fitness function to evaluate the solution (Sayed et al. 2019).
Pathway analysis and protein clustering
The protein–protein interaction between the primary ORAI2 gene, secondary TPI1, and the coalition genes (Table 1) is mapped using the STRING database (Mering et al. 2003). Gene nodes further group the network analysis with k-means clustering of 3 distinct cluster units. In cluster 1, ITPR3 and TRPC6 are grouped, ORAI2, STIM2 in cluster 2, and ITPR1 (Lim et al. 2021), ITPR2, ORAI1, ORAI3, STIM1, TRCP1, and TRPC3 forms cluster 3. All the network genes belong to ITPR, ORAI, STIM, or TRP. Similarly, the TPI1 network is generated with the same number of clusters. ALDOA, ENO1, GAPDH, GAPDHS, GPI, PGK1, PGK2, and TP1 form cluster 1, TALDO1 and TKT belong to cluster 2, and PGM1 alone make cluster 3. The GeneMANIA Cytoscape plug-in (Montojo et al. 2010) predicts the gene function for the root markers ORAI2 and TPI1. The most related gene groups are identified by finding the association with their targets. Figure 2 depicts the interactions between ORAI2 and TPI1 with correlated markers.

The protein–protein interaction between ORAI2, TPI1 and the coalition genes was identified through the genetic algorithm method
Machine learning and modeling
Supervised machine learning classification algorithms are trained with the biomarkers to find the discriminative pattern for classifying the samples of different subgroups. Besides, explainable artificial intelligence techniques such as Local Interpretation and Model Explanations (LIME) and SHapley Additive exPlainer (SHAP) are employed to interpret the model predictions (Lundberg and Lee 2017; Covert et al. 2021). The 250 DEGs identified by statistical analysis and 34 frontal, 60 hippocampal, and 28 temporal subsets identified using the genetic algorithm are used to train ML classifier models. The class label is represented in binary format (0-Case and 1-Control). Logistic Regression (LR), Random Forest (RF), Linear Support Vector Machines (L-SVM), Naive Bayes (NB), and Multilayered Perceptron Neural Network (MLP-NN) algorithms are used in this experiment. The sample size is not evenly distributed in each dataset, so a fivefold cross-validation technique is applied for model validation. The dataset is minimally imbalanced with the target classes. The performance of the trained models is evaluated with accuracy, precision, recall, F1-score, Matthew's correlation coefficient, and receiver's operational characteristic (ROC) curve to avoid evaluation bias.
Explainable artificial intelligence
The essence of XAI is articulated by enunciating the methods and process of machine learning models to human dilettante (Antoniadi et al. 2021). XAI techniques are employed to describe the model predictions, the outcomes, and the biases. The characterization of model transparency, accuracy, and fairness becomes viable with XAI. In this study, the evaluation of the discriminative capacity of ML models is implemented by Local Interpretable Model-Agnostic Explanations (LIME) and SHapley Additive exPlanation (SHAP). These two XAI methods are model-agnostic and post hoc, applied after the model has been trained. LIME interprets ML models' predictions and remains model-independent by perturbing the input data. Observing the resulting impact on the output and the interaction with the local fidelity provides more information on individual predictions. While in the SHAP model, Shapley values explain the individual predictions (Vollert et al. 2021). The technique explains the model output by computing the individual feature contribution. The Shapley values are computed by coalitional game theory (Sekaran and Shanmugam 2022).
Results
The scores of individual ML classifiers attained on candidate markers identified by the genetic algorithm are given (Table 2 and Fig. 3). Table 2 contains the outputs of frontal, hippocampal, and temporal datasets. The results have shown that the performance of NB, L-SVM, MLP-NN, and LR are the same on the frontal, but all five classifiers attained 100% performance on the hippocampal dataset. NB and MLP-NN outperformed other classifiers on the temporal dataset by scoring 100% output. Figure 4 illustrates the accuracy of ML models on DEG and GA subsets. In Fig. 3, the Naive Bayes algorithm stands out from the other classifiers with top performance. Figure 4 represents the performance of ML classifiers on three datasets. The graphs depict the scores of each classifier, and almost all attained better results.

Accuracy of machine learning models on the DEG and GA features of 3 datasets

Performance of machine learning models on sample groups of Alzheimer's dataset
The machine learning classifier's predictions were analyzed in the next phase with LIME and SHAP XAI models. Logistic regression is trained with the GA-identified subsets with 75% training data. In Fig. 5, two samples, each from frontal AD and non-AD, are randomly selected to test the predictions of the trained ML model. The prediction probability indicates the chances of being predicted as either class based on the features. ORAI2, RAB6A, and 7,981,324 are the top three biomarkers identified by LIME to predict frontal AD samples, and 79,814,324, 7,894,213, and COX4I1 for frontal non-AD samples with 0.97 and 0.96 probability respectively. The feature value represents the importance of the particular feature in the predictions. The ranges given in the graph provide the conditions upon which the classifier makes the decision.

LIME interpretation on samples of Alzheimer's disease
Similarly, 8,015,796, 8,063,347, and SYT5 were the robust biomarkers for the hippocampal AD sample prediction with one probability, and IL1RL2, 8,015,796, and PPP1R11 for hippocampal non-AD sample prediction with a probability score of 0.98. ORAI2, the crucial biomarker identified in this study, is an essential factor in predicting the temporal AD and non-AD samples alongside AP1S1, SYT13, and APOBEC3F, 7,892,609, respectively. The prediction probabilities of both classes from these genes were 0.96 and 0.99. Figure 6 represents the top three genes from each dataset. SHAP scores on the datasets reinforce the result attained by LIME. In Fig. 7, the negative gene values positively impact the AD prediction, while positive gene values on non-AD prediction. The negative SHAP value in blue represents AD (labeled as 0), while the positive in red denotes non-AD (labeled as 1) classes on all three datasets. The ORAI2 gene is a decisive factor in classifying AD samples as 0 and the gene 7981324 as 1 on frontal AD and non-AD samples, respectively. Figure 7 shows the SHAP interpretation on hippocampal AD and non-AD samples, identifying 8,015,796 and IL1RL2 (Luís et al. 2022) as the top contributors to the predictions. The ORAI2 gene is found to have higher significance in a temporal dataset of both classes and stands on top of the other genes, as depicted in Fig. 7. This gene influence the predictions of AD and non-AD temporal samples with a higher score alongside the subordinate genes.

Top 3 gene scores of LIME interpretation on group samples of Alzheimer's disease

SHAP interpretation on samples groups of Alzheimer's disease
Discussion
Transcriptomic studies unravel novel insights into a clinical condition. This study is designed in a three-fold pattern (Fig. 1). The gene expression data is split into subgroups by the blood samples collected from three brain regions. Frontal, hippocampal, and temporal classes are grouped with control samples extracted from the same region. The frontal dataset contains 15 AD and 18 non-AD samples; the hippocampal dataset has eight AD and ten non-AD samples, and the temporal dataset with 10 AD and 19 non-AD samples. The most differentially expressed genes are extracted in the first fold. This step is crucial to finding informative genes from large-dimensional feature sets. The second fold identifies the candidate genetic markers from the DEGs with a genetic algorithm. 34 frontal, 60 hippocampal, and 28 temporal genes are strongly associated with AD. ORAI2 (ORAI calcium release-activated calcium modulator 2) is the standard marker in all the candidate subsets (Table 1 and Fig. 2). Recent studies confirm the gene susceptibility of ORAI2 with AD progression (Scremin et al. 2020; Ma et al. 2021). TPI1 is another gene in both hippocampal and temporal markers (Fig. 2 and Table 1). The TPI1 gene encodes instructions for the production of an enzyme known as triosephosphate isomerase 1. This enzyme is involved in glycolysis, a critical energy-producing process. TPI1 was discovered through proteomics analysis to be a novel biomarker for predicting intrahepatic cholangiocarcinoma recurrence (Yu et al. 2020).
Figure 8 visualizes the overlapping genes identified from the candidate gene subsets. The ORAI2 gene is present in all three datasets, and TPI1 is found in frontal and hippocampal datasets. The ORAI2 and TPI1 genes are further analyzed to trace the genetic coalition linked to other AD-related disease markers (Fig. 2). Cluster analysis is performed on similar group genes by the k-means algorithm with 3 clusters. STIM1, TRCP1, and ITPR1 genes directly impact neurodegenerative conditions (Fig. 2). TRP channels are the potential therapeutic targets for Alzheimer's and related illnesses (Yamamoto et al. 2007; Datta et al. 2020; Hwang et al. 2021). Also, mutations in these genes are found to have pathological relevance to AD. TRPC6, a dominant pathogenic gene, is known for the early onset of AD, which is strongly influenced by the mutation caused by APP or PS1 genes (Dahlgren et al. 2002; Dillen and Annaert 2006). The molecular interactions between the ORAI2 and TPI1 are represented as a gene network using the genemania web tool, a Cytoscape plug-in available for association studies (Fig. 2). The biomarkers are mapped to the correlated genes most directly or partially associated with AD. TRPC6, ORAI2, and STIM2 are evident, playing a significant role in AD pathogenesis and the regulation of Store-operated calcium entry (SOCE). These genes also have pharmacological properties in drug discovery activities (Hunanyan et al. 2021).

Overlapping gene ratio plot using Venn diagram
The third fold of this experiment evaluates the prominence of the identified candidate markers using machine-learning models (Fig. 3). Five classification algorithms are trained with the reduced dataset: naive Bayes, linear support vector machines, multilayered perceptron backpropagation, logistic regression, and random forest. The five-fold cross-validation method is chosen to train and validate the model performance (Fig. 3). The model scores are calculated with precision, recall, F1-score, accuracy, and MCC. The DEGs and GA-identified subsets are separately trained. As expected, the scores of DEGs are lower compared to GA markers. The irrelevant features from the DEGs are eliminated during the GA selection process.
The performance of ML models differs in every dataset, with different scores from each model. The naive Bayes algorithm performs better on all the datasets, scoring 90.9%, 96.9%, 94.4%, 100%, 93.1%, and 100% on frontal-DEG's-GA, hippocampal-DEGs-GA, and temporal-DEGs-GA respectively (Table 2). The higher scores of the models strengthen the genetic evidence found as a prominent marker of AD. The increase in performance displays the model's ability to discriminate the case and control AD samples. The dataset is minimally imbalanced over the binary target classes. The most reliable statistical metric, MCC, is calculated to avoid prediction bias by considering all four components of the confusion matrix—true positive, false positive, true negative, and false negative. Besides, precision, recall, and f1-scores ensure the robustness of the model evaluation. Surprisingly, the GA-identified subset performance against all ML classifiers in the hippocampal dataset is 100% in all calculated metrics. This evident finding exhibits the complete association of the genetic markers with AD. In temporal-GA, the NB classifier attained 100% performance in all scoring criteria, outperforming other classification algorithms. Apart from the random forest model, all other classifiers displayed the same performance on the frontal-GA dataset. In an evident study, nine lncRNAs named LncSigAD9 discriminate AD and healthy samples with higher sensitivity and specificity at 86.3% and 89.5%, respectively, with receiver operating characteristic curves of 0.863 (Zhou et al. 2019). A similar study with lncRNA and microRNA analyzed the expression patterns and revealed essential dysregulation genes. The investigations showed 85% to 95% accuracy in biomarker detection with ML models (Garcia-Fonseca et al. 2021).
The interpretation of ML model output discloses the essential genes contributing to the accurate prediction of AD or Non-AD samples. The black-box processing of ML algorithms minimizes visibility and increases uncertainty. Explainable artificial intelligence techniques are built to uncover the ML model's internal processes. This experimental study implements the LIME and SHAP methods to discover the prediction pattern from the ML algorithms trained with the candidate genes. The single observation from both AD and non-AD for all three datasets is fetched randomly for evaluation. The logistic regression model is used to train the datasets with 75% samples from which the explanation for the predictions is extracted. The LR learner is selected because it performs better with binary classification problems. XAI interpretation analysis allows comprehension of the internal process of AI models and makes it easier to interpret the decision-making of AI systems. The algorithm can be retraced, and a high level of visibility minimizes the "black box" effect. The salient feature of XAI is enabling humans to trust the predictions, maintain a high level of performance, and have context-aware decision-making (Pawar et al. 2020). There exist many types of XAI models for disparate objectives. The XAI methods can be either model-specific or model-agnostic, intrinsic or post hoc, and local or global. In this study, the evaluation of the discriminative capacity of ML models is implemented by LIME and SHAP. These two XAI methods are model-agnostic and post hoc, applied after the model has been trained.
LIME identified ORAI2, RAB6A, and probe7981324 as important biomarkers to predict a sample into the frontal AD class. The feature value of ORAI2 is 10.63, which shows a higher priority than other genes. probe7981324, probe7894213, COX4I1 predicts frontal-non-AD. probe8015796, probe8063347, and SYT5 are the predictor markers of hippocampal-AD and IL1RL2, probe8015796, and PPP1R11 for hippocampal non-AD. ORAI2 again stands on top in predicting temporal AD samples alongside AP1S1 and SYT13 genes. ORAI2, APOBEC3F, and probe7892609 genes contribute more to predicting temporal non-AD cases. The SHAP model displayed its depth interpretation of the LR model prediction on different samples. The base value in a SHAP output represents the average predictions made by the model on the training dataset. The output value is the model-predicted value for the sample. For a random sample of temporal AD class, SHAP identified RAB33A influencing the prediction to 1, but ORAI2, RAB6A to 0. This interpretation proves the prediction as accurate that temporal AD is mapped to class 0 and temporal non-AD as 1.
RAB6A positively impacts predicting the sample as 0, but probe7981324, and probe7894213 to class 1, ensuring the genes contributing to the predictions and their closeness. 8,015,796 is more promising for hippocampal AD, and for hippocampal non-AD, IL1RL2 is found as the biomarker. ORAI2 is a common gene involved in predicting both temporal AD and non-AD. The identified biomarkers could act as targeted therapeutics for AD. The XAI models unveil profound insights by mining the prediction patterns of machine learning models. Further studies on the association between genes and disease through XAI techniques deliver promising findings and improve treatment outcomes.
Data availability
The data are available with the corresponding author GPDC.
Code availability
The source code of the implementation is deposited in the GitHub repository. Link: https://github.com/karthiksekaran/alzheimer-biomarker-study-XAI
References
Al-Thani HF, Ahmad MN, Younes S, Zayed H (2021) Genetic variants associated with alzheimer disease in the 22 arab countries: A systematic review. Alzheimer Dis Assoc Disord 35(2):178–186
Antoniadi AM, Du Y, Guendouz Y, Wei L, Mazo C, Becker BA, Mooney C (2021) Current challenges and future opportunities for XAI in machine learning-based clinical decision support systems: a systematic review. Appl Sci 11(11):5088
Bagabir, S., Ibrahim, N. K., Bagabir, H., & Ateeq, R. (2022). Covid-19 and artificial intelligence: Genome sequencing, drug development and vaccine discovery. J Infect Publ Health
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, ... Soboleva A (2012) NCBI GEO: archive for functional genomics data sets—update. Nucl Acids Res 41(D1):D991–D995
Bhattacharyya R, Teves CAF, Long A, Hofert M, Tanzi RE (2022) The neuronal-specific isoform of BIN1 regulates β-secretase cleavage of APP and Aβ generation in a RIN3-dependent manner. Sci Rep 12(1):1–12
Chethana HP, Hemachandra G, Sidhu A (2022) Biomarkers: Potential Perspectives in Detection, Diagnosis, and Prognosis of Neurodegenerative Disorders. In: Elumalai P, Lakshmi S (eds) Functional foods and therapeutic strategies for neurodegenerative disorders. Springer, Singapore, pp 203–222
Covert I, Lundberg SM, Lee SI (2021) Explaining by removing: A unified framework for model explanation. J Mach Learn Res 22:209–211
Dahlgren KN, Manelli AM, Stine WB, Baker LK, Krafft GA, LaDu MJ (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem 277(35):32046–32053
Datta S, Rajnish KN, George Priya Doss C, Melvin Samuel S, Selvarajan E, Zayed H (2020) Enzyme therapy: a forerunner in catalyzing a healthy society? Expert Opin Biol Ther 20(10):1151–1174
Dillen K, Annaert W (2006) A two decade contribution of molecular cell biology to the centennial of Alzheimer’s disease: are we progressing toward therapy? Int Rev Cytol 254:215–300
Doss CGP, Zayed H (2017) Comparative computational assessment of the pathogenicity of mutations in the Aspartoacylase enzyme. Metab Brain Dis 32(6):2105–2118
Garcia-Fonseca A, Martin-Jimenez C, Barreto GE, Pachon AFA, Gonzalez J (2021) The emerging role of long non-coding RNAs and MicroRNAs in neurodegenerative diseases: A perspective of machine learning. Biomolecules 11(8):1132
Gunning D (2019) Aha, DW DARPA’s explainable artificial intelligence (XAI) program. AI Mag 40:44–58
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH (2009). The WEKA data mining software: An update. SIGKDD explorations. 11(1)
Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, Nakabeppu Y (2014) Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex 24(9):2476–2488
Hunanyan L, Ghamaryan V, Makichyan A, Popugaeva E (2021) Computer-based drug design of positive modulators of store-operated calcium channels to prevent synaptic dysfunction in Alzheimer’s disease. Int J Mol Sci 22(24):13618
Hwang SM, Lee JY, Park CK, Kim YH (2021) The role of TRP channels and PMCA in brain disorders: intracellular calcium and pH homeostasis. Front Cell Dev Biol 9:584388
John H (1992) Holland genetic algorithms. Sci Am 267(1):44–50
Koutsodendris N, Nelson MR, Rao A, Huang Y (2022) Apolipoprotein e and Alzheimer’s disease: Findings, hypotheses, and potential mechanisms. Annu Rev Pathol 17:73–99
Kushwaha P, Singh V, Somvanshi P, Bhardwaj T, Barreto GE, Ashraf GM, Mishra BN, Chundawat RS, Haque S (2021) Identification of new BACE1 inhibitors for treating Alzheimer’s disease. J Mol Model 27(2):58
Lim B, Prassas I, Diamandis EP (2021) Alzheimer disease pathogenesis: The role of autoimmunity. J Appl Lab Med 6(3):756–764
Linardatos P, Papastefanopoulos V, Kotsiantis S (2020) Explainable ai: A review of machine learning interpretability methods. Entropy 23(1):18
Luís JP, Simões CJ, Brito RM (2022) The therapeutic prospects of targeting IL-1R1 for the modulation of neuroinflammation in central nervous system disorders. Int J Mol Sci 23(3):1731
Lundberg SM, Lee SI (2017) A unified approach to interpreting model predictions. Adv Neural Inf Process Syst 30
Ma Y, Dammer EB, Felsky D, Duong DM, Klein HU, White CC, Zhou M, Logsdon BA, McCabe C, Xu J, Wang M (2021) Atlas of RNA editing events affecting protein expression in aged and Alzheimer’s disease human brain tissue. Nat Commun 12(1):1–6
Meena J, Hasija Y (2022) Application of explainable artificial intelligence in the identification of Squamous Cell Carcinoma biomarkers. Comput Biol Med 146:105505
Mering CV, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31(1):258–261
Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, ... Bader GD (2010) GeneMANIA Cytoscape plug-in: fast gene function predictions on the desktop. Bioinformatics 26(22):2927–2928
Patel S, Bansoad AV, Singh R, Khatik GL (2022) BACE1: A key regulator in Alzheimer’s disease progression and current development of its inhibitors. Curr Neuropharmacol 20(6):1174–1193
Pawar U, O'Shea D, Rea S, O'Reilly R. Incorporating explainable artificial intelligence (XAI) to aid the understanding of machine learning in the healthcare domain. In AICS 2020 (pp. 169–180)
Sayed S, Nassef M, Badr A, Farag I (2019) A nested genetic algorithm for feature selection in high-dimensional cancer microarray datasets. Expert Syst Appl 121:233–243
Scheubert L, Lustrek M, Schmidt R, Repsilber D, Fuellen G (2012) Tissue-based Alzheimer gene expression markers-comparison of multiple machine learning approaches and investigation of redundancy in small biomarker sets. BMC Bioinformatics 13:266
Scremin E, Agostini M, Leparulo A, Pozzan T, Greotti E, Fasolato C (2020) ORAI2 down-regulation potentiates SOCE and decreases Aβ42 accumulation in human neuroglioma cells. Int J Mol Sci 21(15):5288
Sekaran K, Shanmugam S (2022) Interpreting the factors of employee attrition using explainable AI. In: 2022 International Conference on Decision Aid Sciences and Applications (DASA) (pp. 932–936). IEEE
Sekaran K, Sudha M (2021) Predicting autism spectrum disorder from associative genetic markers of phenotypic groups using machine learning. J Ambient Intell Humaniz Comput 12(3):3257–3270
Sery O, Zeman T, Sheardova K, Vyhnalek M, Markova H, Laczo J, Lochman J, Kralik P, Vrzalova K, Dziedzinska R, Balcar VJ, Hort J (2022) Six genetically linked mutations in the CD36 gene significantly delay the onset of Alzheimer’s disease. Sci Rep 12(1):10994
Sneha P, Ebrahimi EA, Ghazala SA, Kumar DT, Siva R, Doss CGP, Zayed H (2018) Structural analysis of missense mutations in galactokinase 1 (GALK1) leading to galactosemia type-2. J Cell Biochem 119(9):7585–7598
Thirumal Kumar, D., Eldous, H. G., Mahgoub, Z. A., George Priya Doss, C., & Zayed, H. (2018). Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson's and Gaucher diseases. Metab Brain Dis 33(6):1835–1847
Vollert S, Atzmueller M, Theissler A. Interpretable Machine Learning: A brief survey from the predictive maintenance perspective. In:2021 26th IEEE international conference on emerging technologies and factory automation (ETFA) 2021. IEEE, pp 01–08
Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y (2007) Transient receptor potential channels in Alzheimer's disease. Biochim Biophys Acta (BBA)-Mol Basis Dis 1772(8):958–67
Yang Y, Zhou D, Zhang X, Shi Y, Han J, Zhou L, ..., Zhu W (2022) D3AI-CoV: a deep learning platform for predicting drug targets and for virtual screening against COVID-19. Brief Bioinform 23(3):bbac147
Yu WL, Yu G, Dong H, Chen K, Xie J, Yu H, Ji Y, Yang GS, Li AJ, Cong WM, Jin GZ (2020) Proteomics analysis identified TPI1 as a novel biomarker for predicting recurrence of intrahepatic cholangiocarcinoma. J Gastroenterol 55(12):1171–1182
Zafeiris D, Rutella S, Ball GR (2018) An artificial neural network integrated pipeline for biomarker discovery using Alzheimer’s disease as a case study. Comput Struct Biotechnol J 16:77–87
Zaki OK, Krishnamoorthy N, El Abd HS, Harche SA, Mattar RA, Nofal MY, ..., Zayed H (2017) Two patients with Canavan disease and structural modeling of a novel mutation. Metab Brain Dis 32(1):171–177
Zhou M, Zhao H, Wang X, Sun J, Su J (2019) Analysis of long noncoding RNAs highlights region-specific altered expression patterns and diagnostic roles in Alzheimer’s disease. Brief Bioinform 20(2):598–608
Acknowledgements
The authors gratefully acknowledge the Vellore Institute of Technology (VIT) for providing the facilities to carry out this work. Authors Dr. Karthik Sekaran and Dr. George Priya Doss. C also acknowledges the Indian Council of Medical Research (ICMR), the Government of India agency, for the research grants (No. BMI/12(13)/2021, ID No: 2021-6359)
Funding
Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Contributions
KS, HZ, and CGPD contributed to designing the study and data acquisition, analysis, and interpretation. KS and AMA conducted the experiments, which involved the acquisition, analysis, interpretation of the results, and manuscript drafting. CGPD and HZ supervised the entire study and were involved in understanding the data and editing the manuscript. The manuscript was reviewed and approved by all the authors.
Corresponding authors
Ethics declarations
Ethics approval (include appropriate approvals or waivers)
Not Applicable.
Consent to participate (include appropriate statements)
Not Applicable.
Consent for publication (include appropriate statements)
Not Applicable.
Competing interest
The authors have declared that no conflicts of interest exist.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sekaran, K., Alsamman, A.M., George Priya Doss, C. et al. Bioinformatics investigation on blood-based gene expressions of Alzheimer's disease revealed ORAI2 gene biomarker susceptibility: An explainable artificial intelligence-based approach. Metab Brain Dis 38, 1297–1310 (2023). https://doi.org/10.1007/s11011-023-01171-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01171-0