
Abstract
Natural flavonoids such as genistein, kaempferol and daidzein were previously found to be able to reduce efficiency of glycosaminoglycan synthesis in cells of patients suffering from mucopolysaccharidoses, inherited metabolic diseases with often brain disease symptoms. This feature was employed to test these compounds as potential drugs for treatment other neuronopathic lysosomal storage disorders, in which errors in sphingolipid metabolism occur. In this report, on the basis of DNA microarray analyses and quantitative real time PCR experiments, we present evidence that these compounds modify expression of genes coding for enzymes required for metabolism of sphingolipids in human dermal fibroblasts (HDFa). Expression of several genes involved in sphingolipid synthesis was impaired by tested flavonoids. Therefore, it is tempting to speculate that they may be considered as potential drugs in treatment of LSD, in which accumulation of sphingolipids, especially glycosphingolipids, occurs. Nevertheless, further studies on more advances models are required to test this hypothesis and to assess a therapeutic potential for flavonoids in this group of metabolic brain diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sphingolipids are ubiquitous components of mammalian cell membranes. Their derivatives, glycosphingolipids (GSLs), constitute a complex group modified by the addition of a carbohydrate unit. GSLs may be divided in two major sets, represented by glucosylceramides (GlcCers) and galactosylceramides (GalCers). Dominant type of glucosylceramides, i.e. gangliosides are the major glycoconjugates found in the nervous system (Schnaar 2005), while galactosylceramides have a much more restricted distribution and are generally confined to myelin and kidney (Schnaar 2009). The essential role of these macromolecules is defined by the fact that defects in genes encoding enzymes of both biosynthesis and degradation of GSLs result in many severe metabolic disorders.
Inborn errors of GSL biosynthesis are at low frequency and result in lethality with a failure of embryos to develop beyond gastrulation (Yamashita et al. 1999), severe epilepsy syndrome (Simpson et al. 2004), or spastic paraplegia (Boukhris et al. 2013). De novo biosynthesis of GSL competes with their formation by salvage pathways using sphingolipid fragments released from the lysosomal compartments. Depending on the cell type, 50 - 90 % of glycosphingolipids are derived from the salvage pathways (Gillard et al. 1998; Tettamanti et al. 2003).
In contrast to sparse GSL biosynthetic diseases, there is a relatively large group found to be an outcome of inappropriate macromolecule catabolism, caused by inherited failure in genes encoding lysosomal proteins. GSLs are degraded along a strictly sequential pathway in humans, and for almost every degradation step, a disease has been described in which the correlated enzyme or activator protein is defective. Lysosomal accumulation of GSLs occurs predominantly in cells and organs that have the highest rates of biosynthesis or uptake of the undegradable sphingolipids and their precursors (Schultz et al. 2011). Such massive accumulation prevents lysosomal function, blocks signaling cascades dependent on Akt-mTOR and Erk (Takamura et al. 2008), and inhibits nutrition delivery through the endolysosomal system, leading to a state of cellular starvation (Jeyakumar et al. 2009).
GLS degradation process occurs in acidophilic cell compartment, endosomal/lysosomal system. Defects in GLS degradation can lead to lysosomal storage disorders (LSDs). Sphingolipidoses are a subgroup of LSDs involving accumulation of sphingolipids and glycosphingolipids (Kolter and Sandhoff 2006). They all occur through two distinct mechanisms of metabolism, defects in biosynthesis or degradation (Platt et al. 2012). One of several factors contributing to the molecular pathogenesis and clinical form of LSDs is the level of residual catabolic activity. Correlation between functional residual catabolic activity and the progression of the lipid storage disorder (Conzelmann and Sandhoff 1983) was basically confirmed for different clinical forms of diseases such as metachromatic leukodystrophy (Leinekugel et al. 1992; Tan et al. 2010), GM2-gangliosidosis (Leinekugel et al. 1992), Gaucher disease (Gieselmann 2005) and Niemann-Pick type A and B diseases (Ferlinz et al. 1995).
Sphingolipid accumulation also occurs in a group of LSDs in which the primary issue is not a lack of a degradation enzyme. Mostly these diseases are associated with problems with trafficking and fusion in the endocytic system, leading to secondary storage of GSLs (Platt et al. 2012). Niemann-Pick type C (NPC), a disease thought to be mainly a disorder of cholesterol transport from the lysosome is an extreme example of this (Rosenbaum and Maxfield 2011).
Among several ways of treatment for LSDs, most effective is enzyme replacement therapy (ERT) (Brady 2006; Lim-Melia and Kronn 2009). Currently, only seven from more than 50 lysosomal disorders are treated with ERT, namely, Gaucher disease, Fabry disease, Pompe disease and mucopolysaccharidosis types I, II, IVA and VI (Lim-Melia and Kronn 2009; Rohrbach and Clarke 2007; Tomatsu et al. 2014). Although such a recombinant enzyme can be delivered to most tissues and organs, it cannot cross the blood–brain-barrier (BBB), and thus, ERT is not effective in treatment of neurological symptoms. Despite the great progress which has been made over the past 20 years, we still lack satisfactory therapies for lysosomal brain diseases.
Until now, only two of LSDs belonging to sphingolipidosis subgroup, Gaucher disease and Fabry disease, can be treated with ERT (Banikazemi et al. 2007; Pastores et al. 2005; Pastores 2010; Tajima et al. 2009). Moreover, an alternative treatment of Gaucher disease is available, which is based on reduction of the substrate synthesis (Hollak et al. 2009). Besides, it was demonstrated that genistein (5, 7-dihydroxy-3- (4-hydroxyphenyl)-4H-1-benzopyran-4-one) can significantly reduce efficiency of synthesis of glycosaminoglycans (GAGs), compounds that accumulate in mucopolysaccharidoses (MPSs) (Piotrowska et al. 2006). The mechanism of action of genistein is based on inhibition of phosphorylation of the epidermal growth factor receptor (EGFR) (Jakobkiewicz-Banecka et al. 2009). As it was shown previously genistein and other flavonoids have also significant impact on expression pattern of genes coding for lysosome metabolism and macromolecule degradation proteins, by the influence of the TFEB regulatory pathway (Moskot et al. 2014; Sardiello et al. 2009). Effects of flavonoids on GAG synthesis and storage, as well as on global gene expression, reported recently (Moskot et al. 2015), revealed that these compounds caused effective reduction in levels of GAG production in fibroblasts, and induced remarkable alterations in profiles of transcripts derived from genes coding for enzymes involved in GAG metabolism and from many other genes. Transcriptomic analyses indicated that, despite certain similarities, there are significant differences between effects of the tested flavonoids (genistein, kaempferol and daidzein used alone, and in combinations) on global gene expression patterns in cells. Moreover, these studies revealed that among the tested compounds, genistein (an isoflavone), kaempferol (a flavonol) and a mixture of these two flavonoids had the most pronounced effects on the regulation of genes’ activities. Interestingly, effects of the mixture of genistein and kaempferol were stronger that those revealed by any of these flavonoids used alone (Moskot et al. 2015). It is worth to mention that these flavonoids can cross the BBB (Tsai 2005), what makes reasonable considering these compounds as potentially useful in the optimization of treatment for neuronopathic forms of LSDs, including sphingolipidoses. Therefore, the aim of this work was to assess effects of flavonoids on expression of genes involved in sphingolipid metabolism.
Materials and methods
Cell cultures of fibroblasts
Cell line of Human Dermal Fibroblasts (HDFa) was obtained from the Cascade Biologics, Portland, OR, USA. Fibroblasts were cultured from cryo-preserved cells in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (FBS) and 1 % antibiotic/antimycotic solution (Sigma-Aldrich, MO, USA) at 37 °C in a humidified atmosphere of 5 % CO2. Genistein was synthetized at the Pharmaceutical Research Institute (Warsaw, Poland), while kaempferol and daidzein were obtained from Sigma-Aldrich (Steinheim, Germany). Tested flavonoids were dissolved in dimethyl sulfoxide (DMSO) and added to the indicated final concentrations, as determined in previous studies (Kloska et al. 2011; Piotrowska et al. 2006), to cell cultures. For experimental procedures, cells were plated to a confluence of approximately 80 %. After overnight incubation, culture medium was replaced with fresh medium either flavonoid-free one, containing DMSO at a final concentration of 0.05 % (control, Ctrl), or the one supplemented with appropriate amounts of tested flavonoids. The experimental treatment was carried out for 1, 24 and 48 h period.
RNA extraction
Total RNA was extracted from cells using the High Pure RNA Isolation Kit (Roche Applied Science, Indianapolis, USA) following the manufacturer’s instructions. The quality and quantity of each RNA sample was evaluated using the RNA 6000 Nano Assay on the Agilent 2100 Bioanalyser (Agilent Technologies Inc., USA).
Microarray assays for mRNA analysis
Abundance of various mRNAs was measured using Illumina’s Human HT-12 v3 Expression BeadChips (Illumina Inc., CA, USA) [http://www.illumina.com/Documents/products/datasheets/datasheet_humanht_12.pdf]. The BeadChip supports highly efficient whole-genome transcriptional studies targeting more than 25,000 genes with more than 48,000 probes. Probes were designed using the RefSeq and the UniGene databases. Illumina TotalPrep RNA Amplification Kit (Ambion, TX, USA) was used in order to amplify total input RNA. Following linear amplification by T7 RNA polymerase, in vitro synthesized, biotin-labeled cRNA was applied to the BeadChip for hybridization according to the manufacturer’s instructions. BeadChips were scanned using an Illumina BeadArray Reader and the Bead Scan Software (Illumina Inc., CA, USA). The quality of microarray data was controlled by examining raw and adjusted intensity histograms. The detection scores (detection p value) were used to determine expressions of each gene using BeadStudio software 2.3.4 (Illumina Inc., CA, USA), which was computed based on Z-values of a gene relative to the Z-value of negative controls on expression chip. Normalization of gene expression data were done by the GeneSping software 7.3.1 (Agilent Technologies, CA, USA) as the value normalized per chip and per gene with median.
Data extraction and statistical analysis
Illumina’s Human HT-12v4 Expression BeadChips (Illumina Inc., USA) were used for the microarray analysis of three biological replicates. The assay performance, data extraction and statistical analysis were performed as previously described (Moskot et al. 2014). Gene expression data have been deposited in the NCBI’s Gene Expression Omnibus (GEO, http:www.ncbi.nlm.nih.gov/geo, GEO Series accession number GSE43692 and GSE34074) according to the MIAME standards. The comparison of relative fold change of gene expression levels between flavonoid-treated and non-treated cells was performed. Sphingolipid metabolism pathways were defined according to the KEGG annotation (Kanehisa et al. 2006) (Path: hsa00600, hsa00601, hsa00603 and hsa00604) and AmiGO (GO:0030148, GO:0006665 and GO:0030149).
Quantitative real-time RT-PCR for mRNA analysis
Quantitative real-time Reverse Transcription PCR (real-time qRT-PCR) was performed to measure the mRNA levels of the studied genes using the LightCycler® System 480 (Roche Applied Science, Indianapolis, USA). Total RNA was reverse-transcribed into cDNA using Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, IN, USA), according to the manufacturer’s instructions. Quantification of mRNA levels was conducted using LightCycler TaqMan Master (Roche Applied Science, IN, USA). In case of ARSA, ASAH1, HEXA, NEU1, NPC1 and SUMF1 genes real-time qRT-PCR analysis was performed using Real Time ready Custom Panel (cat no. 05532914001, config. no. 90014731, Roche Applied Science, Indianapolis, USA) and LightCyclerH 480 Probes Master (Roche Applied Science, Indianapolis, USA), as determined in previous study (Moskot et al. 2015). However, primers utilized for specific amplification of the CLN8, GBA1, GLA, GM2A, HEXB, NPC2, PPT1 and SMPD1 genes were obtained from Thermo Fisher Scientific GmbH. The 2-∆∆ct method was used to determine the relative gene transcript levels after normalization to the reference gene coding for glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Ref1), TATA box binding protein (TBP, Ref2), phosphoglycerate kinase 1 (PGK1, Ref1), and transferrin receptor (TFRC, Ref2). Primers and probes of reference genes GAPDH (NM_002046), PGK1 (NM_000291), TBP (NM_003194.4) and TFRC (ENST00000392396) were bought from Roche Applied Science, IN, USA. Negative controls were tested in parallel. Primers, probes and assay ID are described in Table 1.
Results
Profiling of expression of genes involved in sphingolipid metabolism with microarrays
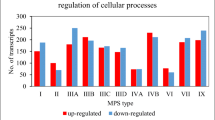
Microarray analysis of transcripts derived from genes coding for sphingolipid metabolism enzymes showed those which expression was identified as different by at least 0.7 and 1.3 fold change (p < 0.05) following flavonoids’ exposure in relation to control of non-treated samples of human dermal fibroblasts (HDFa). As discovered in five independent assays, most compound-dependent changes among 121 genes of GSL metabolism occurred at 100 μM kaempferol-treated cells for 48 h (i.e. 29, with 19 up- and 10 down-regulated genes) as shown in Fig. 1. For 24 h treatment with particular flavonoids, 27 genes (i.e. 18 and 9 with a greater than 2-fold increase and decrease in expression, respectively) had modulated expression for 100 μM kaempferol, 20 revealed up-regulation for 100 μM genistein, while 9 were down-regulated for 100 μM kaempferol. After 48 h of exposition to various flavonoids, we found 21 transcripts to be remarkably up-regulated by genistein and kaempferol mix treatment, while ten were found to be down-regulated by 100 μM kaempferol.

Number of genes revealing altered expression in response to various flavonoids’ treatment type (genistein, kaempferol, daidzein, and mixtures of them as stated), identified in the microarray analysis of GSL metabolism associated transcripts (0.7 ≥ FC ≥ 1.3) of HDFa cells, with the p-value <0.05, and n ≥ 3
Studying carefully the gene expression levels modulated after 24 h of treatment with tested flavonoids, we found one transcript (i.e. PPAP2A) in common between all tested conditions, while 5 mRNAs were up-regulated after 48 h of incubation with flavonoids (i.e. CL4A3BP, NEU1, PPAP2A, SMPDL3A and SUMF1). PPAP2A mRNA was therefore included in both 24 and 48 h time course sets. Considering particular flavonoids for both 24 and 48 h treatment, among genes with positively regulated activity, we found 14 for 100 μM genistein (i.e. ARSA, ASAH1, COL4A3BP, GBA1, GBGNT1, GM2A, HEXA, KDSR, NEU1, NPC1, PPAP2A, SMPD1, SMPDL3A and SUMF1), 13 for 100 μM kaempferol (i.e. CLN8, GLA, HEXA, KDSR, LARGE, NEU1, NPC1, PPAP2A, SMPDL3A, SPHK1, SPHK2, ST3GAL1 and SUMF1), 5 for genistein and kaempferol mixture (i.e. GBA1, GBGT1, PPAP2A, SMPDL3A and SUMF1), 3 for 100 μM daidzein (i.e. ASAH1, NEU1 and PPAP2A), and 3 for genistein and daidzein mixture (i.e. GBGT1, NEU1 and PPAP2A). Taking on account genes with a decreased expression, five transcripts were found to be modulated after both 24 and 48 h of treatment with 100 μM kaempferol (i.e. B3GNT1, ELOVL1, SPTLC2, ST3GAL6 and ST6GALNAC5), while one for genistein and kaempferol mix (i.e. ST3GAL6).
When looking at studied conditions, i.e. at least in one experimental state we found several transcripts with increased and/or decreased expression, belonging to various regulation modules with corresponding overlap of some genes between the sets of regulatory processes (Tables 2 and 3). Among those genes we determined several transcripts (i.e. ARSA, ASAH1, CLN8, GALC, GBA1, GLA, GM2A, HEXA, HEXB, NAGA, NEU1, NPC1, NPC2, PPT1, SMPD1 and SUMF1) being associated with sphingolipids’ disorders (Table 4).
Real-time qRT-PCR analysis of expression of genes involved in sphingolipid metabolism
Real-time quantitative Reverse Transcription PCR (real-time qRT-PCR) was used to confirm a set of gene expression changes observed in the microarray analysis. To choose the most stable genes as internal references for real-time qRT-PCR data normalization, two candidates - GAPDH (Ref1) and TBP (Ref2) for CLN8, GBA1, GLA, GM2A, HEXB, NPC2, PPT1 and SMPD1, while other two - PGK1 (Ref1) and TFRC (Ref2) for ARSA, ASAH1, HEXA, NEU1, NPC1, and SUMF1 genes, respectively, were selected according to their expression levels detected in the microarray studies. From 59 GSL metabolism genes regulated under tested conditions (Table 3), 14 from 16 genes coding for enzymes involved in glycosphingolipid metabolism disorders (as shown in Table 4) were experimentally validated (Fig. 2). For the two other genes, GALC and NAGA designing of intron spanning primers was not possible, therefore they were not included in the analysis. Basically, the real-time qRT-PCR and microarray methods showed a good agreement on regulated genes (Fig. 2).

Effect of genistein (100 μM, a), kaempferol (100 μM, b), and mix of genistein and kaempferol (30 μM each, c) on mRNA level of selected genes up-regulated with an expression ratio greater than 1.3-fold, and with statistical significance set at p ≤ 0.05, involved in sphingolipid metabolism in HDFa cells after 24 h of treatment. Two types of analyses were made: microarray gene expression studies (white columns) and real-time qRT-PCRs (light and dark grey columns), where the data represent averaged values ±SD from n ≥ 3 and denote significant differences for samples treated with tested compound/s against non-treated samples, with respect to reference genes of constant expression level: GAPDH (Ref 1) and TBP (Ref 2) for CLN8, GBA1, GLA, GM2A, HEXB, NPC2, PPT1 and SMPD1, while PGK1 (Ref 1) and TFRC (Ref 2) for ARSA, ASAH1, HEXA, NEU1, NPC1, and SUMF1, respectively. *Results based on raw data from Moskot et al. (2015)
Discussion
Since molecular mechanisms of LSDs are rather well understood (relative to many other diseases of this nature), and as they were among the first genetic diseases for which effective - at least to some extent - treatment became possible, these disorders may be considered as models in inherited metabolic diseases. There are several LSDs, in which accumulation and storage of sphingolipids occur as the primary cause of symptoms (Sandhoff 2013).
In the light of problems with crossing BBB, it appears crucial to look for potential drugs that could be used in pathological sphingolipid storage. One group of such potential drugs are flavonoids, such as genistein (an isoflavone), kaempferol (a flavonol) and daidzein (an isoflavone), compounds which were reported previously to partially inhibit GAG synthesis and to reduce GAG storage in fibroblasts derived from patients suffering from mucopolysaccharidoses, metabolic disorders belonging to LSDs (Arfi et al. 2010; Kloska et al. 2011). Since potentially many genes involved in synthesis of various biologically active molecules could be controlled by the EGF-dependent pathways, and at the same time some other transcripts regulating lysosomal administration are regulated by the TFEB pathway, we asked if previously selected and studied flavonoids can also influence expression of genes coding for enzymes required for sphingolipid metabolism.
In this report, we demonstrate that flavonoids influence expression of dozens of genes involved in sphingolipid metabolism. In general, the DNA microarray analysis indicated that kaempferol, genistein and mix of these two compounds profiled the expression of the greatest number of genes (Fig. 1). When looking at particular studied conditions, we found numerous of transcripts with altered expression belonging to various regulation modules, such as: GSL biosynthesis - globo series, GSL biosynthesis - lacto and neolacto series, GSL biosynthesis - ganglio series, lactosylceramide biosynthesis, glycerolipid metabolism, ceramide biosynthesis, sphingosine biosynthesis, sphingosine degradation, other sphingolipid metabolism genes, intracellular trafficking or fusion GSL catabolic enzyme modification, with corresponding overlap of genes between these sets of regulatory processes (Table 2). Additionally, we conclude that 59 among 121 genes of GSL metabolism were regulated at least in one experimental condition (Table 3). What is interesting, 16 genes from them are associated with well-known sphingolipids disorders (Table 4).
Previous studies indicated that flavonoids, particularly genistein and kaempferol, modified expression of genes coding for metabolism of GAGs in a specific manner. Namely, expression of many (but not all) genes which products are involved in synthesis of GAGs were down-regulated while a few were up-regulated, whereas vast majority of transcripts derived from genes coding for GAG-degradation enzymes were more abundant after treatment with genistein, kaempferol or their mixture (Moskot et al. 2014). In the case of GSL metabolism, the picture is more complicated. In both groups, up- and down-regulated by flavonoids, there are genes coding for enzymes involved in synthesis and degradation of particular lipids’ derivatives. Moreover, in each particular pathway for production and degradation of specific class of GSLs, there are always stimulated and inhibited genes coding for enzymes operating in both opposite pathways. One might predict that inhibition of a crucial step in synthesis of particular GSL may results in impairment of the whole process, even if other steps are stimulates, according to the bottleneck mechanisms, i.e. the restriction of the efficiency of the whole pathway by the slowest reaction. However, such a hypothesis requires experimental verification.
It does not escape our attention that fibroblasts are not the best models for genetic and biochemical changes in neurons, while diseases caused by GSLs’ accumulation cause brain dysfunctions. However, for in vitro experiments with the microarray technology, which allowed us to monitor expression of many genes at the same time but which also require relatively large amount of biochemically good quality material isolated from actively growing cells, human fibroblasts appeared to be the optimal choice. Other possible experimental systems would have severe restrictions in the availability of the material and viability of cells. Nevertheless, it is obvious that any further studies which might lead to determination if flavonoids can be considered as potential drugs for management of GSL-storage diseases should involve either cultures of cells types which are models more closely resembling physiology of the brain or animal models for particular diseases.
In summary, our results indicate that expression of several genes involved in sphingolipid synthesis can be regulated by tested flavonoids. Therefore, it is tempting to speculate that they may be considered as potential drugs in treatment of LSD, in which accumulation of sphingolipids, especially glycosphingolipids, occurs. Nevertheless, further studies on more advances models are required to test this hypothesis and to assess a therapeutic potential of flavonoids in this group of metabolic brain diseases.
References
Arfi A, Richard M, Gandolphe C, Scherman D (2010) Storage correction in cells of patients suffering from mucopolysaccharidoses types IIIA and VII after treatment with genistein and other isoflavones. J Inherit Metab Dis 33:61–67
Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, Mcdonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ (2007) Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 146:77–86
Boukhris A, Schule R, Loureiro JL, Lourenco CM, Mundwiller E, Gonzalez MA, Charles P, Gauthier J, Rekik I, Acosta Lebrigio RF, Gaussen M, Speziani F, Ferbert A, Feki I, Caballero-Oteyza A, Dionne-Laporte A, Amri M, Noreau A, Forlani S, Cruz VT, Mochel F, Coutinho P, Dion P, Mhiri C, Schols L, Pouget J, Darios F, Rouleau GA, Marques Jr W, Brice A, Durr A, Zuchner S, Stevanin G (2013) Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet 93:118–123
Brady RO (2006) Enzyme replacement for lysosomal diseases. Annu Rev Med 57:283–296
Conzelmann E, Sandhoff K (1983) Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci 6:58–71
Ferlinz K, Hurwitz R, Weiler M, Suzuki K, Sandhoff K, Vanier MT (1995) Molecular analysis of the acid sphingomyelinase deficiency in a family with an intermediate form of Niemann-Pick disease. Am J Hum Genet 56:1343–1349
Gieselmann V (2005) What can cell biology tell us about heterogeneity in lysosomal storage diseases? Acta Paediatr Suppl 94:80–86 discussion 79
Gillard BK, Clement RG, Marcus DM (1998) Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology 8:885–890
Hollak CE, De Fost M, Van Dussen L, Vom Dahl S, Aerts JM (2009) Enzyme therapy for the treatment of type 1 gaucher disease: clinical outcomes and dose - response relationships. Expert Opin Pharmacother 10:2641–2652
Jakobkiewicz-Banecka J, Piotrowska E, Narajczyk M, Baranska S, Wegrzyn G (2009) Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J Biomed Sci 16:26
Jeyakumar M, Williams I, Smith D, Cox TM, Platt FM (2009) Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol Dis 34:406–416
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34:D354–D357
Kloska A, Jakobkiewicz-Banecka J, Narajczyk M, Banecka-Majkutewicz Z, Wegrzyn G (2011) Effects of flavonoids on glycosaminoglycan synthesis: implications for substrate reduction therapy in Sanfilippo disease and other mucopolysaccharidoses. Metab Brain Dis 26:1–8
Kolter T, Sandhoff K (2006) Sphingolipid metabolism diseases. Biochim Biophys Acta 1758:2057–2079
Leinekugel P, Michel S, Conzelmann E, Sandhoff K (1992) Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet 88:513–523
Lim-Melia ER, Kronn DF (2009) Current enzyme replacement therapy for the treatment of lysosomal storage diseases. Pediatr Ann 38:448–455
Moskot M, Montefusco S, Jakobkiewicz-Banecka J, Mozolewski P, Wegrzyn A, Di Bernardo D, Wegrzyn G, Medina DL, Ballabio A, Gabig-Ciminska M (2014) The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J Biol Chem 289:17054–17069
Moskot M, Jakobkiewicz-Banecka J, Kloska A, Smolinska E, Mozolewski P, Malinowska M, Rychlowski M, Banecki B, Wegrzyn G, Gabig-Ciminska M (2015) Modulation of expression of genes involved in glycosaminoglycan metabolism and lysosome biogenesis by flavonoids. Sci Rep 5:9378
Pastores GM (2010) Recombinant glucocerebrosidase (imiglucerase) as a therapy for Gaucher disease. BioDrugs 24:41–47
Pastores GM, Barnett NL, Kolodny EH (2005) An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. Clin Ther 27:1215–1227
Piotrowska E, Jakobkiewicz-Banecka J, Baranska S, Tylki-Szymanska A, Czartoryska B, Wegrzyn A, Wegrzyn G (2006) Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur J Hum Genet 14:846–852
Platt FM, Boland B, Van Der Spoel AC (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199:723–734
Rohrbach M, Clarke JT (2007) Treatment of lysosomal storage disorders : progress with enzyme replacement therapy. Drugs 67:2697–2716
Rosenbaum AI, Maxfield FR (2011) Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J Neurochem 116:789–795
Sandhoff K (2013) Metabolic and cellular bases of sphingolipidoses. Biochem Soc Trans 41:1562–1568
Sardiello M, Palmieri M, Di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325:473–477
Schnaar RL (2005) In: Fukuda M, Rutishauser U, Schnaar RL, Yamaguchi Y (eds) Neuroglycobiology. Oxford University Press, Oxford, pp. 95–113
Schnaar RL (2009) Essentials of glycobiology. Cold Spring Harbour Laboratory Press, Cold Spring Harbour
Schultz ML, Tecedor L, Chang M, Davidson BL (2011) Clarifying lysosomal storage diseases. Trends Neurosci 34:401–410
Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, Wang H, Wiznitzer M, Gurtz K, Verganelaki A, Pryde A, Patton MA, Dwek RA, Butters TD, Platt FM, Crosby AH (2004) Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet 36:1225–1229
Tajima Y, Kawashima I, Tsukimura T, Sugawara K, Kuroda M, Suzuki T, Togawa T, Chiba Y, Jigami Y, Ohno K, Fukushige T, Kanekura T, Itoh K, Ohashi T, Sakuraba H (2009) Use of a modified alpha-N-acetylgalactosaminidase in the development of enzyme replacement therapy for Fabry disease. Am J Hum Genet 85:569–580
Takamura A, Higaki K, Kajimaki K, Otsuka S, Ninomiya H, Matsuda J, Ohno K, Suzuki Y, Nanba E (2008) Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun 367:616–622
Tan MA, Fuller M, Zabidi-Hussin ZA, Hopwood JJ, Meikle PJ (2010) Biochemical profiling to predict disease severity in metachromatic leukodystrophy. Mol Genet Metab 99:142–148
Tettamanti G, Bassi R, Viani P, Riboni L (2003) Salvage pathways in glycosphingolipid metabolism. Biochimie 85:423–437
Tomatsu S, Almeciga-Diaz CJ, Montano AM, Yabe H, Tanaka A, Dung VC, Giugliani R, Kubaski F, Mason RW, Yasuda E, Sawamoto K, Mackenzie W, Suzuki Y, Orii KE, Barrera LA, Sly WS, Orii T (2014) Therapies for the bone in mucopolysaccharidoses. Mol Genet Metab
Tsai TH (2005) Concurrent measurement of unbound genistein in the blood, brain and bile of anesthetized rats using microdialysis and its pharmacokinetic application. J Chromatogr A 1073:317–322
Yamashita T, Wada R, Sasaki T, Deng C, Bierfreund U, Sandhoff K, Proia RL (1999) A vital role for glycosphingolipid synthesis during development and differentiation. Proc Natl Acad Sci U S A 96:9142–9147
Acknowledgments
This work was supported by National Science Centre (project grant no. UMO-2011/01/B/NZ1/03686 and N N301 668540).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s11011-017-9974-7.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Moskot, M., Jakóbkiewicz-Banecka, J., Smolińska, E. et al. Activities of genes controlling sphingolipid metabolism in human fibroblasts treated with flavonoids. Metab Brain Dis 30, 1257–1267 (2015). https://doi.org/10.1007/s11011-015-9705-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-015-9705-x