
Abstract
Glutamate excitotoxicity has been implicated as one of the pathological mechanisms contributing to neuronal cell death and is involved in many neurological disorders. Stem cell transplantation is a promising approach for the treatment of nervous system damage or diseases. Previous studies have shown that mesenchymal stem cells (MSCs) have important therapeutic effects in experimental animal and preclinical disease model of central nervous system pathology. However, it is not well understood whether neurogenesis of MSCs or MSC conditioned-medium (CM) containing microparticles mediates therapeutic effects. Here, we investigated the neuroprotective effects of human adipose-derived MSCs (AMSCs) on cortical neurons using models of glutamate excitotoxicity. Following exposure to glutamate (100 μM, 15 min), cortical neurons were co-cultured with either AMSCs separated by a semiporous membrane (prohibiting direct cell-cell contact) or with AMSC-CM for 18 h. Compared to untreated control groups, AMSCs and AMSC-CM partially and similarly reduced neuronal cell damages, as indicated by reduced LDH release, a decreased number of trypan-positive cells and a decline in the number of apoptotic nuclei. Protection by CM was associated with increased GAP-43 expression and an elevated number of GAP-43-positive neurites. Furthermore, CM increased levels of ATP, NAD+ and NADH and the ratio of NAD+/NADH, while preventing a glutamate-induced decline in mitochondrial membrane potential. These results demonstrate that AMSC-CM mediates direct neuroprotection by inhibiting neuronal cell damage/apoptosis, promoting nerve regeneration and repair, and restoring bioenergy following energy depletion caused by glutamate excitotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Excitotoxicity is defined as cell death resulting from the toxic action of excitatory amino acids. Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS) and it is involved in many physiological functions including development of the nervous system, learning and memory. However, excessive and prolonged exposure to glutamate under pathological circumstances can lead to overstimulation of glutamate receptors and excitotoxic cell death (Lau and Tymianski 2010). Glutamate excitotoxicity is implicated as a common pathological mechanism contributing to neuronal cell injury in a number of neurological disorders as it plays a critical role in the pathogenesis of stroke, epilepsy, brain trauma and neurodegenerative diseases (Lipton and Rosenberg 1994; Dong et al. 2009).
It is clear that the glutamate-induced neuronal death is mediated by the entry of extracellular Ca2+ as a result of the activation of N-Methyl-D-aspartic acid (NMDA) subtype of glutamate receptors and a resultant calcium overload (Arundine and Tymianski 2003). Excessive intracellular calcium triggers pathways leading to cell death. Indeed, it has been suggested that intracellular calcium overload may result in mitochondrial dysfunction, energy metabolism disorder, oxidative stress and apoptosis. Recent studies have suggested that mitochondrial dysfunction and subsequent energetic collapse are critical steps in the progression to cell death in glutamate excitotoxicity (Nicholls 2004; Smaili et al. 2011).
Bioenergy homeostasis is foundational for maintaining normal cell function and survival. Neurons are excitable cells that require a large amount of energy to maintain membrane potential and synaptic transmission, making the brain the most energy -consuming organ in the body. Thus, neurons are extremely susceptible to bioenergetic stress. Irreversible neuronal death occurs if the brain is deprived of oxygen for more than 5 min. Among many neural activities, the excitatory glutamatergic systems use the most energy (Sibson et al. 1998; Shen et al. 1999; Raichle and Gusnard 2002). The energy consumption of neurons increases following activation by glutamate. Energy metabolism disorder is involved in multiple acute and chronic neurological diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, stroke and brain trauma (Mattson et al. 1999; Ferreira et al. 2010; Amato and Man 2011; Mochel et al. 2012). Reduced cell energy supply is one of the early and primary pathological events in these diseases. Furthermore, under these pathological conditions, neural energy depletion is accompanied by a massive release of glutamate and excitotoxic cell injury (Del Rio et al. 2007; Nicholls et al. 2007). Hence, minimizing glutamate-induced neuronal cell injury and rescuing glutamate-induced neuronal energy depletion is a direct and effective therapeutic approach.
One repair strategy following nerve injury is to promote axonal regeneration or outgrowth. In recent years, several molecules have been found associated with axonal regeneration, and among them the most well-known is the growth-associated protein 43 (GAP-43) (Frey et al. 2000). GAP-43, which was discovered in the early 1980s, is a membrane-bound protein expressed mainly in the growth cores, axons and presynaptic terminals of neurons (Benowitz et al. 1987). It is particularly abundant in periods of neurite outgrowth during development and regeneration in the central and peripheral nervous systems. GAP-43 is closely related to neural development, axonal regeneration and synaptic reorganization, and is regarded as an intrinsic factor during development and regeneration (Benowitz and Routtenberg 1997). Therefore, GAP-43 is considered as an axonal regeneration marker (Aoki et al. 2007) and a neural restoration marker (Lopatina et al. 2011).
As the capacity of the CNS to regenerate after acute or chronic lesions is limited, there are currently no effective therapeutic strategies for many neurological diseases. Recently, however, many studies indicate that stem cell transplantation is a promising approach for the treatment of nervous system damages or diseases. Somatic stem cells, especially neural stem cells and mesenchymal stem cells (MSCs), have demonstrated the character to reduce neurological defects and promote functional recovery in experimental animal models of CNS pathology (Hofstetter et al. 2002; Zhang et al. 2006; Ohtaki et al. 2008) and in preclinical trials for stroke and multiple sclerosis (Bang et al. 2005; Freedman et al. 2010; Martino et al. 2010).
The mechanisms by which MSCs provide neuroprotection under CNS pathological conditions are not well known. Early research showed that MSCs could differentiate into CNS glial cells and neurons in vitro and in vivo (Kopen et al. 1999; Sanchez-Ramos 2002). But it is controversial whether MSCs are able to functionally replace damaged neural cells besides expressed neuronal or glial markers (Phinney and Prockop 2007). There is evidence that MSCs stimulate endogenous protective and restorative responses via paracrine mechanisms (Uccelli et al. 2008). Recent studies on MSCs secretome suggest that MSCs secrete numerous bioactive factors that mediate neuroprotection via trophic support, immunomodulation, anti-apoptosis, anti-fibrosis and angiogenesis (Singer and Caplan 2011; Skalnikova et al. 2011). MSCs were found to protect against glutamate-induced apoptosis in rat pheochromocytoma cell line PC12 cells through the secretion of neurotrophic factors (Lu et al. 2011). Growth factors such as BDNF and bFGF protected cerebellar granular neurons against glutamate excitotoxicity by increasing glucose reuptake and stabilizing intracellular Ca2+ and the mitochondrial electrochemical gradient (El Idrissi and Trenkner 1999). IGF-1 was demonstrated to reverse a decrease in GAP-43 expression induced by glutamate in dorsal root ganglion neurons (Liu et al. 2012). Adult adipose-derived MSCs (AMSCs) are particularly attractive for use in cell transplantation as they can be obtained and expanded in vitro with a relative ease. However, it is still unknown whether neurogenesis of MSCs or MSC-CM containing rich microparticles can prevent glutamate-induced energy depletion in neurons.
In the present study, in vitro models of glutamate excitotoxicity were used to investigate whether AMSC-CM mediate neuroprotection in cultured cortical neurons by regulating GAP-43 expression and energy metabolism, in order to provide an experimental basis for further exploration of AMSCs use for clinical applications.
Experimental procedures
Cell culture
Culture and identification of human AMSCs
Human adipose tissue (lipoaspirate) from healthy 35 to 45 year-old donors was obtained by liposuction procedures under anesthesia. All donors were informed and gave their written consent with procedures approved by the Ethics Committee on the Use of Human Subjects (Dalian Medical University and affiliated hospitals). AMSCs were isolated as previously described (Zuk et al. 2002). In brief, adipose tissue was washed in phosphate-buffered saline (PBS) and digested with 0.1 % collagenase type І (sigma) in PBS for 30 min at 37 °C in a water-bath shaker. The digested tissue was centrifuged for 5 min at 1,000 rpm at room temperature and the supernatant containing mature adipocytes, debris and connective tissue was aspirated. The pellet was resuspended and plated in T25 culture flasks in Dulbecco’s modified Eagle medium (DMEM; supplemented with D-glucose 4,500 mg/l, 4 mM L-glutamine and 110 mg/l sodium pyruvate; GIBCO, Invitrogen), 10 % fetal bovine serum (FBS; GIBCO, Invitrogen) and 100 U penicillin/100 μg streptomycin (GIBCO, Invitrogen) at a density of 2 × 106/cm2. Cultures were maintained at 37 °C in a humidified atmosphere of 95 % O2 and 5 % CO2. After reaching 80–100 % confluence, cells were passaged using 0.25 % trypsin/0.38 % EDTA (GIBCO, Invitrogen) and kept in medium as described above. All experiments were performed with cells passages 4–6.
AMSCs were routinely characterized by flow cytometry analysis using antibodies to the surface markers CD13, CD34, CD106 (PE-conjugated mouse anti-human; BD Biosciences) and CD44, CD45, CD90 (FITC-conjugated mouse anti-human; BD Biosciences). AMSCs were positive (>95 %) for CD13, CD44 and CD90 and negative (<5 %) for CD34, CD45 and CD106, indicating their mesenchymal nature. AMSCs differentiation into adipocytes and osteoblasts was performed as follows. AMSCs were seeded at a density of 105/ml in 35 mm dishes and grown in DMEM growth medium. After reaching 80–100 % confluence, mediums were replaced with adipocyte or osteoblast induction mediums (Hyclone). After 14 days of differentiation, adipocytes were stained with Oil Red O (Sigma) and fresh red lipid vacuoles were observed in the cytoplasm of nearly all AMSCs. After 28 days of osteoblast differentiation, cells were stained with Von Kossa and showed the mineralized matrix deposition.
Primary neuron culture
All protocols were performed in accordance with the guidelines of the Animal Care Committee of Dalian Medical University. Primary neuronal cultures were obtained from cerebral cortices of neonatal Sprague–Dawley rats (Laboratory Animal Center of Dalian Medical University) as described previously (Brewer 1997; Beaudoin et al. 2012). Briefly, tissue was digested in DMEM/F-12 (GIBCO, Invitrogen) with 2 mg/ml papain (Worthington) and 2 U/ml DNase I (Worthington) at 37 °C for 30 min and dissociated by mild trituration in DMEM/F12 containing 10 % FBS (GIBCO, Invitrogen) and 10 % donor equine serum (Hyclone). For survival analysis and immunocytochemistry, 5–7 × 105/ml dissociated cells were plated in 24-well plates or coverslips previously coated with poly-D-lysine (PDL; Sigma). For biochemical procedures, 1–2 × 106/ml cells were seeded in six-well plates coated with PDL. Cultures were maintained at 37 °C in a humidified atmosphere of 95 % O2 and 5 % CO2. After 4 h, mediums were replaced and cells were maintained with neurobasal medium (Invitrogen) supplemented with 2 % B-27 supplement (PAA Laboratories) and 0.5 mM glutamine. Half of the medium was refreshed every 3 days. All experimental treatments were performed in vitro on day 9 (DIV9). Immunofluorescence staining with anti-microtubule associated protein-2 (MAP-2; Sigma) antibody and anti-glial fibrillary acidic protein (GFAP; Millipore Chemicon) antibody revealed that cultures contained more than 90 % neurons.
Preparation of conditioned medium from AMSCs
AMSCs at passages 4–6 were seeded on 35 mm culture dishes with DMEM at a density of 105/ml. After 48 h, the confluent cells were washed once with PBS and then incubated in neurobasal medium containing B-27 supplement. After another 18–20 h, the medium was collected and designated as conditioned medium (CM).
Experimental treatments
Excitotoxic death was induced in DIV9 rat cortical neurons by exposure to different concentrations of glutamate (10, 50 and 100 μM) supplemented with 10 μM glycine (assist agonist of NMDA receptors) for 15 min at 37 °C. The medium was then replaced with either the original culture medium or different concentrations of AMSC-CM ranging from 10 to 100 % (v/v) and cells continued to be cultured for 18 h. Control groups were treated under the same conditions but in absence of glutamate and glycine.
Co-culture of AMSCs and neurons
Following exposure to glutamate for 15 min, neurons were co-cultured with AMSCs. AMSCs were separated from neurons with a porous membrane (0.4 μm) which allowed for the exchange of molecules but prohibited cell-cell direct contact. Cortical neurons were cultured in 12- or 6-well plates for 9 days in vitro prior to excitotoxicity. AMSCs were plated in 12- or 6-well inserts (polyester, 1.12 cm2; Corning) and grown to 80–100 % confluence in serum-containing growth medium. Prior to placing AMSCs-containing inserts into plates containing neurons, AMSCs were rinsed twice with PBS and medium was changed to neuronal medium, consequently removing serum from the co-culture. Cells were co-cultured for 18 h and then neural damages were assessed.
Assessments of cell damages
After cortical neurons were cultured in 24-well plates for 9 days, 100 μM glutamate and 10 μM glycine was added and cultured for 15 min at 37 °C. The medium was then replaced with either the original culture medium or the optimum concentration of CM, or inserts containing AMSCs were added to damaged neurons. After 18 h, cell damages were evaluated by trypan blue dye (Sigma) and lactate dehydrogenase (LDH) release. Trypan blue dye was performed as previously described with modification. 18 h after insult, culture medium was removed and replaced with 0.12 mg/ml trypan blue dissolved in PBS. Cells were incubated at room temperature for 10 min and washed with PBS. Cultures were then fixed with 4 % paraformaldehyde for 30 min and cells were visualized under a phase-contrast microscope. Total numbers of dead and intact neurons were counted and expressed as a percentage of the total number of cells. For statistical purposes, the average of ten neighbor fields for each condition was used as a single data. LDH was assayed using the Cytotoxicity Detection Kit plus (Roche Applied Science) according to the manufacturer’s instructions. Results were read on an EL808 microplate assay reader (Gene Company Limited). Untreated cells were used for control groups and the culture medium without cells was used as background control. Data represent the percentage relative to control treated with lysis buffer. Each group contained three samples and experiments were repeated at least three times.
TUNEL and Nissl Staining
An In Situ Cell Death Detection Kit (Fluorescein, Roche Applied Science) based on the TUNEL assay was used to evaluate apoptotic cell death following the manufacturer’s instructions. For negative controls, reactions were performed by omitting TUNEL enzyme TdT and cells were incubated with the label solution provided in the kit. No reactivity was observed when TdT was absent. Cells were stained with fluorescent Nissl dye (TRITC, Molecular Probe) to label the total neurons and were observed with a fluorescence microscope (LEICA, DFC500). The number of TUNEL-positive cells and Nissl-positive cells were counted in at least three separate experiments per treatment condition without knowledge of treatment history.
Immunofluorescent labeling of GAP-43
Briefly, cultured neurons were fixed with 4 % paraformaldehyde for 30 min and permeabilized with 0.3 % Triton X-100 for 20 min at room temperature. After blocking unspecific binding sites with 10 % bovine serum albumin (BSA; Sigma), cells were incubated overnight with monoclonal rabbit anti-GAP-43 (1:200; Abcam) antibody at 4 °C. Cells were then washed for three times in 0.01 M PBS and incubated with specific secondary antibody conjugated FITC (1:100; goat anti-rabbit; Sigma) for 60 min at room temperature. For negative controls, reactions were performed by omitting the primary antibody. No reactivity was observed when the primary antibody was absent. Nuclei were counterstained with Hoechst33258 (10 μg/ml; Sigma). Cells were observed and photographed with a fluorescence microscope (LEICA, DFC500).
Western blotting analysis for detection of the protein levels of GAP-43
Protein levels of GAP-43 under different experiment conditions were detected by Western blot assay with β-actin as an internal control. Cultured neurons in six-well plates were washed three times with cold PBS and lysed on ice with RIPA buffer containing protease inhibitor for 30 min. Samples were centrifuged at 12,000 rpm for 20 min at 4 °C and the supernatant was collected for Western blot assays. Protein levels in the supernatant were determined using a BCA kit (Beyotime Biotechnology). 35 μg of protein from each sample was loaded on 12 % SDS-PAGE gels. After separation by electrophoresis, proteins were transferred to PVDF membranes (Millipore) and blocked unspecific binding with 5 % non-fat dry milk. Membranes were incubated with rabbit anti-GAP-43 monoclonal antibody (1:500; Abcam) overnight at 4 °C. Following three washes with TBST, the blots were incubated with goat anti-rabbit IgG-HRP (1:1,000) for 1 h at room temperature. Immunoreactive bands were visualized with an ECL detection kit (Thermo) on light sensitive film. Bands were analyzed with Gel-Pro Analyze4 software. Data represent the percentage relative to the control group.
Measurement of ATP levels
Total intracellular ATP concentrations in neurons were measured with a luciferin-luciferase assay-based commercial kit (Bioassay). Briefly, neurons grown in 6-well plates were washed three times with cold PBS and then lysis buffer was added to each well. Cells were collected after thoroughly scraping the culture at the well bottom. Cell lysates were centrifuged at 12,000 rpm for 20 min. The supernatant was used for ATP assay. Luminescence was measured using a luminometer (Corning). A standard curve and equation were generated using an ATP standard and used to calculate ATP concentrations in samples.
Measurement of NAD+ and NADH levels
Total intracellular NAD+/NADH concentrations in neurons were measured using an EnzychromTM NAD+/NADH assay kit (Bioassay). The assay is based on an enzyme-catalyzed kinetic reaction. Briefly, NAD+/NADH was extracted from cells at 60 °C for 5 min, neutralized with the opposite extraction and then centrifuged at 14,000 rpm for 5 min. NAD+ extracts in the supernatant were converted to NADH by enzymatic cycling with lactate dehydrogenase, which reduces MTT to formazan. Optical density was measured at 560 nm using a plate reader (Gene Company Limited). The intensity of the reduced produce color is proportional to the NAD+/NADH concentration in the sample. A standard curve and equation were generated using an NAD standard and used to calculate NAD+/NADH concentrations in samples.
Determination of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was evaluated with the fluorescent probe tetramethylrhodamine ethyl ester (TMRE, Invitrogen) using methods described previously. Cells were loaded with 10nM TMRE at 37 °C for 30 min, washed, and maintained in culture medium throughout experiments. Images of TMRE fluorescence were captured with a phase-contrast fluorescent microscope (LEICA, DFC500) at excitation and emission wavelengths of 488 nm and 525 nm respectively. The mean intensity of fluorescence was measured and calculated using Image-Pro Plus 6.0 software.
Statistical analysis
Statistical analysis was performed using SPSS 11.5. Datas are presented as mean±SEM. The difference between mean values was determined by one-way ANOVA. P values of less than 0.05 were considered statistically significant.
Results
AMSC-CM attenuated neuronal damages in a concentration-dependent manner
To examine the glutamate excitotoxicity on neurons, DIV9 primary cultured cortical neurons were treated with glutamate (10–100 μM) supplemented with 10 μM glycine for 15 min and neurobasal medium was replaced and maintained for 18 h. Cell damage was measured by LDH release. The results showed that glutamate exposure displayed evident toxicity in cultured cortical neurons and led to cell damage in a dose-dependent manner (Fig. 1a). 50 μM glutamate induced a modest but significant LDH release (19.48 ± 0.77 %). More extensive damage occurred with 100 μM glutamate (40.26 ± 0.60 %). 100 μM glutamate supplemented with 10 μM glycine was used as an excitotoxicity model for subsequent experiments because of LDH release from nearly half of the cells at this dose.

Glutamate was toxic to cultured cortical neurons and AMSC-CM reduced glutamate-induced neuronal damage in a concentration-dependent manner. a Cortical neurons at DIV9 were exposed to glutamate (10–100 μM) supplemented with 10 μM glycine for 15 min and the original culture medium was replaced. Cell damage was determined by LDH release after 18 h. Glutamate induced a dose-dependent neuronal damage. 100 μM glutamate supplemented with 10 μM glycine was chosen for all subsequent experiments. b Different concentrations of CM were added to damaged neurons following glutamate exposure and cell damage was evaluated by LDH release after 18 h. Note that glutamate-induced neuron injury was attenuated most effectively by 50 % CM, whereas 100 % CM did not mediate any neuroprotective effects. As the optimal concentration of CM, 50 % CM was used for all subsequent experiments. Data are presented as mean±SD. *** P < 0.001 versus control group, # P < 0.05, ## P < 0.01, ### P < 0.001 versus glutamate treated group. One-Way ANOVA
In order to investigate whether AMSC-CM could protect neurons against glutamate excitotoxicity, different concentrations of CM (10–100 %) were added to damaged neurons caused by 15 min of exposure to glutamate. Cells were incubated for another 18 h. The neuroprotective effect of CM was evaluated by LDH release. As shown in Fig. 1b and Table 1, CM reduced glutamate-induced neuronal injury in a concentration- dependent manner with a maximum protective effect at 50 % CM. At CM contents lower than 50 % or at 70 % CM, protective effects were less pronounced. Note that 100 % CM (all medium replaced with CM) did not mediate any protection. 50 % CM was used for all subsequent experiments because of the optimal neuroprotection (lowest LDH release).
AMSCs and CM protected cortical neurons against glutamate-induced damages and apoptosis
To investigate whether AMSCs could mediate neuroprotection, conditioned medium or a transwell co-culture system, in which cortical neurons were co-cultured in indirect contact with AMSCs and separated by a semiporous membrane (pore size 0.4 μm) allowing for the exchange of small molecules, were used. As shown in Fig. 2a, glutamate induced cortical neurons death as measured 18 h post-incubation by LDH release and trypan blue assay. However, when neurons were co-cultured with AMSCs or CM, LDH release and the number of trypan-positive cells significantly decreased compared with glutamate treated group. But comparing with uninjured controls, cell damage was only partially preserved, indicating a limitation of the protective effect of AMSCs on injured neurons. Furthermore, there was no significant difference in cell damage between neurons co-cultured with AMSCs and those incubated with CM (P > 0.05 in all cases).

AMSCs reduced glutamate-induced damage and apoptosis in cortical neurons. Following 15 min of glutamate treatment, neurons were co-cultured with AMSCs or medium was replaced with AMSC-CM. Cell damages and apoptosis were determined by LDH release, trypan blue dye and TUNEL staining after 18 h. a Quantitative analysis of trypan blue dye and the result of LDH release. b Representative photomicrographs of total neurons stained with Nissl (red) and apoptotic nuclei stained with TUNEL (green) and merged photos were shown. (a–c) untreated control group; (d–f) neurons treated with glutamate alone; (g–i) damaged neurons co-cultured with AMSCs; (j–l) glutamate-treated neurons in the presence of AMSC-CM. c Quantitative analyses of Nissl and TUNEL co-staining. Data are presented as mean±SD. *** P < 0.001 versus control group, # P < 0.05, ## P < 0.01, ### P < 0.001 versus glutamate treated group. One-Way ANOVA. Scar bar 200 μm
To further explore whether AMSCs could reduce glutamate-induced neuron apoptosis, TUNEL and Nissl co-staining were performed. A low number of apoptotic cells was seen in the control group (9.80 ± 2.12 %, Fig. 2b [a–c]). The percentage of apoptotic cells increased dramatically in the glutamate group (52.36 ± 2.8 %, Fig. 2b [d–f]). However, apoptosis was suppressed in the AMSCs co-culture (34.32 ± 3.3 %, Fig. 2b [g–i]) and CM groups (30.08 ± 3.5 %, Fig. 2b [j–l]). The reduced percentage of apoptotic cells was still significantly higher (P < 0.05) than those in the uninjured control group (Fig. 3c). Similarly, no significant difference was seen in the percentage of apoptotic cells between neurons co-cultured with AMSCs and those incubated with CM.

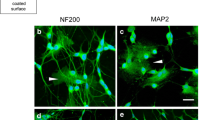
AMSCs restored GAP-43 expression and distribution in cortical neurons. After exposure to glutamate for 15 min, GAP-43 expression and distribution in neurons were measured by immunofluorescence labeling and Western blot. a Representative photomicrographs of GAP-43 (a) control group; (b) glutamate treated group; (c) AMSCs co-culture group; (d) AMSC-CM group. b GAP-43 levels were analyzed using Western blot. Protein loading was checked by stripping and re-probing the membrane with anti-actin antibody. Data are presented as mean±SD. * P < 0.05 versus control group, # P < 0.05, ## P < 0.01 versus glutamate treated group. One-Way ANOVA. Scar bar 200 μm
AMSCs and CM rescued GAP-43 expression and distribution in cortical neurons
To test the effects of glutamate on GAP-43 expression and distribution within cortical neurons, neurons were processed for GAP-43 immunofluorescent labeling and levels of GAP-43 were detected by Western blot. In the control group, GAP-43 was mainly distributed in neuronal bodies and processes. The fluorescence intensity of GAP-43 was homogeneously spread throughout the cytoplasm with weaker dotted distribution in the processes (Fig. 3a[a]). However, in glutamate treated group, GAP-43 intensity decreased markedly and GAP-43 was located neuronal bodies with little expression in processes (Fig. 3a[b]). AMSCs or CM caused an increase in GAP-43 fluorescence intensity and restored typical GAP-43 distribution in the presence of glutamate as compared with the glutamate-only treated group. The GAP-43 fluorescent signal was strong and the distribution in the cytoplasm remained homogeneous. The number of GAP-43-positive neurites increased in the co-culture and CM groups (Fig. 3a[c, d]). The results of Western blot analysis showed that glutamate reduced the level of GAP-43 protein compared with control group (Fig. 3b, 65.18 ± 12.96 %). GAP-43 expression increased markedly when damaged neurons were co-cultured with AMSCs (Fig. 3b, 91.08 ± 2.46 %) or CM (Fig. 3b, 111.62 ± 3.74 %). There was no significant difference (P > 0.05) in GAP-43 expression between the three experimental groups.
AMSCs and CM prevented glutamate-induced reduction in ATP concentration
To examine the effect of glutamate on neuronal bioenergy homeostasis, a luciferase -based ATP assay was used to evaluate total ATP levels in cortical neurons. As a control, a standard curve showed a linear relationship between ATP concentration and fluorescence intensity (Fig. 4a). 100 μM glutamate induced a drastic and significant total ATP reduction (Fig. 4b, 9.07 ± 0.06 μM for control group, 2.89 ± 0.09 μM for glutamate group). AMSCs or CM restored partially the decrease in ATP levels after glutamate exposure (Fig. 4b, 5.28 ± 0.19 μM for co-culture group, 5.66 ± 0.42 μM for CM group).

Glutamate caused a reduction in ATP concentration in cultured cortical neurons. AMSCs or CM prevented glutamate-induced ATP depletion. a A standard curve from an ATP assay showing a linear relationship between ATP levels and luminometer measurement values. b Total cellular ATP concentrations were evaluated with a luciferase-based ATP assay. Data are presented as mean±SD. *** P < 0.001 compared to control group, ## P < 0.01, ### P < 0.001 compared to glutamate treated group. One-Way ANOVA
AMSCs and CM prevented NAD+ and NADH depletion and increased the ratio of NAD+/NADH
Following excitotoxic insult, total cellular NAD+ and NADH levels were measured using an enzymatic cyclic assay. When cortical neurons were exposed to glutamate (100 μM) in combination with glycine (10 μM), a significant decrease of total cellular NAD+ (Fig. 5a, 6.65 ± 0.41 μM for control group, 1.99 ± 0.07 μM for glutamate group) and NADH levels (Fig. 5b, 0.64 ± 0.01 μM for control group, 0.36 ± 0.01 μM for glutamate group) occurred within 18 h. Noticed that ratio of NAD+/NADH was also reduced following glutamate exposure (Fig. 5c, 10.36 ± 0.51 % for control group, 5.48 ± 0.39 % for glutamate group). When neurons were co-cultured with AMSCs or incubated with CM for 18 h, cellular NAD+ and NADH levels sharply increased to 4.60 ± 0.12 μM and 0.54 ± 0.04 μM in the co-culture group, respectively, and 4.42 ± 0.06 μM and 0.46 ± 0.05 μM in the CM group, respectively. The ratio of NAD+/NADH rose to 8.51 ± 0.48 % or 9.70 ± 1.09 % compared to the glutamate treated group.

Glutamate resulted in a reduction in levels of NAD+ and NADH and a decrease in the ratio of NAD+/NADH in cortical neurons. AMSCs or CM prevented NAD+ and NADH depletion. a and b Total cellular NAD+ and NADH levels were measured with an enzymatic cycling assay. c Total cellular ratio of NAD+/NADH. Data are presented as mean±SD. *** P < 0.001 compared to control group,## P < 0.01,### P < 0.001 compared to glutamate treated group. One-Way ANOVA
AMSCs and CM reversed glutamate-induced MMP collapse
To test the impact of glutamate on mitochondrial function, changes in MMP in cultured cortical neurons were estimated using the fluorescent cationic dye TMRE, which preferentially labels active mitochondria based on the highly negative MMP. As shown in Fig. 6, glutamate caused a sharp reduction in active mitochondria (47.74 ± 8.42 % for control group). However, AMSCs or CM treatment resulted in a relative increase in active mitochondria compared to the glutamate treated group (73.0 ± 7.46 % and 63.2 ± 2.33 %, respectively).

Glutamate reduced MMP in cultured cortical neurons. AMSCs or CM preserved MMP collapse following glutamate treatment. MMP was observed using a TMRE fluorescent probe. Image Pro-Plus 6.0 software was used for quantitative TMRE fluorescent density analysis. Data are presented as mean ± SD. *** P < 0.001 compared to control group, # P < 0.05, ## P < 0.01 compared to glutamate treated group. One-Way ANOVA
Discussion
Glutamate excitotoxicity is caused by a massive influx of calcium into neurons due to overstimulation of glutamate receptors and is closely related to neuronal cell injury in stroke, trauma and other chronic neurodegenerative diseases (Lipton and Rosenberg 1994; Lau and Tymianski 2010). In the present study, an in vitro model of glutamate excitotoxicity was used to investigate the neuroprotective effects of MSCs-CM on cortical neurons. The results indicate that AMSCs-CM are able to protect cortical neurons against glutamate excitotoxicity and give the first evidence that AMSCs-CM act by regulating GAP-43 expression and rescuing energy depletion and mitochondrial function.
Our understanding of how the nervous system repair occurs through somatic stem cells, particularly MSCs, has recently changed. MSCs appear to exert their therapeutic effects not because of their intrinsic ‘stemness’, but due to their capacity to release therapeutic molecules that can interact with the host environment (Prockop 2007). Indeed, studies of the MSCs secretome using high performance liquid chromatography-mass spectrometry (HPLC-MS) and antibody-related technologies have demonstrated that MSCs can secrete many kinds of neuro-regulatory molecules, cytokines, growth factors and chemokines which show neuroprotecive and neurorestorative effects, including increasing neuronal viability, inducing the proliferation and differentiation of endogenous neural stem/progenitor cells and promoting regeneration of nerve fibers at sites of injured (Salgado et al. 2009; Singer and Caplan 2011; Skalnikova et al. 2011). The co-culture system used in the present study allowed for the separation of AMSCs from neurons with a semiporous membrane, which prevented direct cell-cell contact and showed that trophic support plays an important role in protecting damaged neurons. In order to further evident the effects of trophic support, AMSC-CM was used for its ability to reduce neural cells injury. The findings reported here show that the neuroprotective effects of AMSC co-culture and CM are not significantly different and both prevent neuronal injury induced by glutamate excitotoxicity in the same extent. This suggests that the observed beneficial effects of AMSCs are mediated by soluble factors secreted by these AMSCs in their conditioned medium. Previous research supports this conclusion. Lu et al. found that MSCs protected PC12 cells from glutamate-induced apoptosis by secreting VEGF, HGF, BDNF and NGF, and neutralization antibody of these factors weakened the protective function of MSCs (Lu et al. 2011). Furthermore, in a recent paper Voulgari-Kokota et al. removed cell-derived microparticles from freshly-prepared MSC-CM using a step-wise centrifugation protocol and found that microvesicle-depleted CM had similar neuroprotective effects against NMDA as complete CM in cortical neurons (Voulgari-Kokota et al. 2012). These finding further indicates that soluble factors secreted by MSCs are what provide neuroprotection against neuronal injury. This is consistent with the view that effects of MSCs are not dependent on their fully integration into the damaged organs after transplantation.
One potential advantage of cell-based therapy to treat neurological disorders is the ability of transplanted cells to sense environmental factors related to tissue damages and response to the injured organs. Previous study has shown that trophic factors released by MSCs increase when they are co-cultured with the injured brain tissue extract (Chen et al. 2002). Isele et al. showed that the protective effects of MSCs on rat neurons against staurosporine (STS) or amyloid-beta peptide-induced apoptosis increased significantly if MSCs were first exposed to neurobasal medium from apoptotic neurons (Isele et al. 2007). But in the present study, remarkable differences in neuron damages were not found between AMSCs co-culture and CM treatment, indicating that any cross-talking between AMSCs and damaged neurons was not detectable under this system. The impact of an injured environment on the ability of MSCs to repair neural cells requires further investigation.
It is important to note that AMSC-CM reduced neural injury induced by glutamate excitotoxicity in a concentration-dependent manner, with an optimal protective effect at 50 % CM. At CM concentrations lower than 50 % or 70 %, the protective effect was diminished. Pure CM (100 %), however, did not lend any neuroprotection. Similarly, Isele NB et al. showed that 100 % BMSC-CM appeared to accelerate STS-mediated apoptosis (Isele et al. 2007). Other researchers have shown that AMSCs can release chemokine and pro-inflammatory factors that are detrimental to damaged tissues (Kilroy et al. 2007; Skalnikova et al. 2011). Adverse effects from MSCs may hasten neuronal death and increase tissue lesions, particularly when MSCs accumulate at the site of lesions. Recent observations demonstrated that intra-cerebroventricular transplantation of MSCs in mice with severe experimental autoimmune encephalomyelitis (EAE), an experimental model of multiple sclerosis, can induce the formation of cellular masses with collagen/fibronectin deposition in the brain parenchyma (Grigoriadis et al. 2011). Previous studies also indicated that MSCs migrated to and accumulated in brain lesion areas after systemically administered MSCs (Chen et al. 2001). However, Liu et al. had reviewed exosomes, a kind of microparticles secreted from cells and without immunogenicity to targeted cells, might be a potential therapeutic vector for metabolic brain diseases (Liu et al. 2013). Thus, microparticles from the AMSC-CM might mediate neuroprotective effects, reduce the toxic substance of conditioned medium and thus avoid the risk of stem cells differentiation into neoplasms.
The foremost regenerative targets following the CNS injury include increasing cell viability, promoting regeneration of axon and myelin and reorganizing synapses. GAP-43 is an intracellular growth-associated protein which appears to assist neuronal path-finding and branching during development and regeneration (Benowitz and Routtenberg 1997). In adults, GAP-43 induces neurotransmitter release, endocytosis and synaptic vesicle recycling by changing the presynaptic membrane. It is involved in neuronal differentiation, plasticity, axonal growth and regeneration and synaptic reconstruction after nerve injury. In vivo and in vitro experiments have demonstrated that GAP-43 plays an important role in the processes of regeneration following central and peripheral nervous system damage. Increased expression of GAP-43 in lesion areas after brain ischemia or trauma was related to the activation of endogenous repair mechanism and the promotion of axonal regeneration in the injured nervous system (Gianola and Rossi 2004). Additionally, GAP-43 was shown to trigger a significant increase in the regeneration of dorsal root ganglion (DRG) axons in adult mice after spinal cord injury in vivo (Bareyre et al. 2002). In the present study, the expression and distribution of GAP-43 after glutamate exposure and the effect of AMSCs on axonal regeneration was observed. The result showed the expression of GAP-43 and the number of GAP-43 positive neurites decreased significantly following glutamate treatment. These indicators increased dramatically to the normal levels when injured neurons were treated with AMSCs or CM, suggesting that AMSCs are able to promote axonal regeneration or outgrowth. The mechanisms may be correlated with neurotrophins or neuro-regulatory factors released by AMSCs which act as positive guidance molecules for axonal growth cone, inducing neurites sprouting and growth following injury. Previous work demonstrated that AMSCs transplantation induced peripheral nerve repair and activated nerve sprout growth in vivo and this ability of AMSCs depended on BDNF secretion (Lopatina et al. 2011). Earlier data indicated that activation of NMDA receptors suppressed GAP-43 expression and axonal outgrowth in hippocampal slice cultures and IGF-1 elevated GAP-43 expression (McKinney et al. 1999). Recently (Li et al. 2013) and (Liu et al. 2012). demonstrated that growth factors neuregulin-1β and IGF-1 could partially reverse decreased GAP-43 expression induced by glutamate in DRG neurons and these effects were involved in activation of PI3K/Akt and ERK1/2 signaling pathways. These reports suggest that AMSCs are able to secrete some soluble factors that activate endogenous restorative and survival mechanisms, increase GAP-43 expression and promote axonal regeneration after nerve injury.
Another important aspect of the present study is that AMSC-CM can reverse the energy depletion induced by glutamate exitotoxicity, as indicated by enhanced MMP, increased ATP, NAD+ and NADH concentrations, and elevated ratio of NAD+/NADH. Increasing evidences suggest that mitochondrial dysfunction play a vital role in glutamate excitotoxicity (Nicholls 2004; Nicholls et al. 2007). Recently, mitochondria have been recognized as the key organelle determining the fate of cells because of their central functions such as ATP synthesis, Ca2+ accumulation, superoxide generation and detoxification and storage of pro-apoptotic proteins (Orrenius 2004; Smaili et al. 2011). MMP is the key parameter controlling these mitochondrial functions, which are of great relevance to neuronal survival. When glutamate-induced elevation of cytoplasmic free calcium ([Ca2+]c) exceeds the mitochondrial Ca2+ set-point at which the organelle behaves as a buffer of [Ca2+]c, mitochondrial Ca2+ overloading occurs (Kiedrowski 1999). Consequently, it leads to decline of MMP and the opening of permeability transition pore (PTP) which disrupts the proton gradient across the inner mitochondrial membrane and causes the bioenergetic collapse and cell death. Several studies indicate that glutamate decreases the ATP concentration in neurons (De Cristobal et al. 2002; Parihar et al. 2008; Foo et al. 2012). The reason for this may be falling of ATP production or increasing of ATP consumption or both. It has been shown that glutamate continues to reduce ATP concentrations even after mitochondrial oxidative phosphorylation and ATP genesis is blocked, indicating that ATP depletion caused by glutamate is associated with increased ATP consumption (Foo et al. 2012). Other data showed a decrease in ATP production following glutamate excitotoxicity (Parihar et al. 2008). Glutamate reduces MMP contributing to a loss of the proton gradient across the mitochondrial membrane, uncoupling of the electron transport chain, inhibition of oxidative phosphorylation and reduction of ATP production. During excitotoxic cell injury, impaired oxidative phosphorylation results in the release of NAD+ from mitochondria, which is finally cleaved by glycohydrolases (Belenky et al. 2007; Soane et al. 2007). It is widely accepted that NAD+ is not only an important energy substrate and cofactor involved in many metabolic reactions (Bieganowski and Brenner 2004; Belenky et al. 2007), but acts as an adenosine donor and source of high energy phosphate for ATP synthesis (Sheline et al. 2000). NAD+ depletion inhibits glycolysis, the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation, all of which reduce ATP levels and lead to energy depletion in neurons (Sheline et al. 2000). Therefore, neuronal NAD+ bioenergetic state and NAD+/NADH redox state are pivotal factors for cell survival after excitotoxic insults (Liu et al. 2008, 2009).
Growth factors such as bFGF, BDNF and taurine have been shown to protect cerebellar granule neurons against excitotoxicity by increasing the glucose uptake and stabilizing the cytoplasmic calcium and mitochondrial electrochemical activity (El Idrissi and Trenkner 1999). A recent study reported that BMSCs genetically modified by Akt elevated ratios of phosphocreatine/ATP and increased phosphocreatine levels in surviving myocardial cells (Gnecchi et al. 2009). More recent data shows that MSC-mediated protection against glutamate excitotoxicity involves the reduction of glutamatergic signaling (Voulgari-Kokota et al. 2012). Changes in gene expression from an activity-dependent mature neuron to a more immature one that is associated with survival and regeneration plasticity, may be a cause of neuronal desensitization to glutamate. Fu H et al. by analyzing neuronal mitochondrial proteomics found that glutamate altered the expression patterns of mitochondrial proteins involved in energy metabolism (electron transport chain, TCA cycle and glucose metabolism), oxidative stress and apoptosis (Fu et al. 2007). Tacrine reversed the expression pattern of these proteins, as well as the decline in MMP, ATP production and neuronal cell death following glutamate treatment. Based on these results and those of previous studies, we surmise that the protective effect of AMSCs against glutamate excitotoxicity observed in the present study might be due to AMSCs themselves or bioactive molecules secreted by them changing the gene expression associated with energy metabolism in neurons, maintaining normal mitochondrial functions and regulating mitochondrial-associated apoptotic signaling pathways. However, the exact mechanisms of MSC-mediated benefit in energy depletion during excitotoxicity need further be explored.
In conclusion, we report that AMSC-CM is able to protect cortical neurons against damage and apoptosis caused by glutamate excitotoxicity, ameliorate a decrease in both GAP-43 expression and the number of GAP-43 positive neurites and prevent glutamate-induced energy depletion. The neuroprotective effects of AMSCs may be related to the release of soluble factors. These findings may provide a new stem cell application method for the treatment of nervous system diseases. But a detailed understanding of the mechanisms of MSC-mediated benefits within the CNS and the identification of the soluble factors mediating MSCs protection will be necessary for further development of the clinical application of MSC-based therapy for neurological disorders.
Abbreviations
- CNS:
-
Central nervous system
- NMDA:
-
N-Methyl-D-aspartic acid
- GAP-43:
-
Growth associated protein-43
- MSCs:
-
Mesenchymal stem cells
- AMSCs:
-
Adipose-derived mesenchymal stem cells
- CM:
-
Conditioned medium
- BDNF:
-
Brain derived neurotrophic factor
- bFGF:
-
Basic fibroblast growth factor
- IGF-1:
-
Insulin-like growth factor
- VEGF:
-
Vascular endothelial growth factor
- HGF:
-
Hepatocyte growth factor
- NGF:
-
Nerve growth factor
- DMEM:
-
Dulbecco’s modified Eagle medium
- FBS:
-
Fetal bovine serum
- EDTA:
-
Ethylenediaminetetraacetic acid
- PDL:
-
Poly-D-lysine
- PBS:
-
Phosphate-buffered saline
- LDH:
-
Lactate dehydrogenase
- TUNEL:
-
TdT-mediated dUTP nick-end labeling
- PVDF:
-
Polyvinylidene fluoride
- SDS:
-
Sodium dodecyl sulfate
- NAD+ :
-
Nicotinamide adenosine denucleotide
- NADH:
-
Nicotinamide adenine denucleotide hydrogen
- TMRE:
-
Tetramethylrhodamine ethyl ester
- ATP:
-
Adenosine triphosphate
- MMP:
-
Mitochondrial membrane potential
- PTP:
-
Permeability transition pore
- TCA:
-
Tricarboxylic acid
References
Amato S, Man HY (2011) Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 10:3452–3460 (Georgetown, Tex)
Aoki YAH, Takahashi K, Miyamoto K, Lenz ME, Moriya H, Masuda K (2007) Axonal growth potential of lumbar dorsal root ganglion neurons in an organ culture system: response of nerve growth factor-sensitive neurons to neuronal injury and an inflammatory cytokine. Spine 32:857–863
Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34:325–337
Bang OY, Lee JS, Lee PH, Lee G (2005) Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol 57:874–882
Bareyre FM, Haudenschild B, Schwab ME (2002) Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci: Off J Soc Neurosci 22:7097–7110
Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7:1741–1754
Belenky P, Bogan KL, Brenner C (2007) NAD+metabolism in health and disease. Trends Biochem Sci 32:12–19
Benowitz LI, Routtenberg A (1997) GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20:84–91
Benowitz LI, Perrone-Bizzozero NI, Finklestein SP (1987) Molecular properties of the growth-associated protein GAP-43 (B-50). Journal Neurochem 48:1640–1647
Bieganowski P, Brenner C (2004) Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+in fungi and humans. Cell 117:495–502
Brewer GJ (1997) Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 71:143–155
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M (2001) Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke: J cereb Circ 32:2682–2688
Chen X, Li Y, Wang L, Katakowski M, Zhang L, Chen J, Xu Y, Gautam SC, Chopp M (2002) Ischemic rat brain extracts induce human marrow stromal cell growth factor production. Neuropathology: Off J Jpn Soc Neuropathol 22:275–279
De Cristobal J, Cardenas A, Lizasoain I, Leza JC, Fernandez-Tome P, Lorenzo P, Moro MA (2002) Inhibition of glutamate release via recovery of ATP levels accounts for a neuroprotective effect of aspirin in rat cortical neurons exposed to oxygen-glucose deprivation. Stroke: J Cereb Circ 33:261–267
Del Rio P, Montiel T, Chagoya V, Massieu L (2007) Exacerbation of excitotoxic neuronal death induced during mitochondrial inhibition in vivo: relation to energy imbalance or ATP depletion? Neuroscience 146:1561–1570
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30:379–387
El Idrissi A, Trenkner E (1999) Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 19:9459–9468
Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF (2010) Multiple defects in energy metabolism in Alzheimer’s disease. Curr Drug Targets 11:1193–1206
Foo K, Blumenthal L, Man HY (2012) Regulation of neuronal bioenergy homeostasis by glutamate. Neurochem Int 61:389–396
Freedman MS, Bar-Or A, Atkins HL, Karussis D, Frassoni F, Lazarus H, Scolding N, Slavin S, Le Blanc K, Uccelli A (2010) The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the international MSCT study group. Mult Scler 16:503–510
Frey DLT, Xu L, Schneider C, Caroni P (2000) Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol 149:1443–1454
Fu H, Li W, Liu Y, Lao Y, Liu W, Chen C, Yu H, Lee NT, Chang DC, Li P, Pang Y, Tsim KW, Li M, Han Y (2007) Mitochondrial proteomic analysis and characterization of the intracellular mechanisms of bis(7)-tacrine in protecting against glutamate-induced excitotoxicity in primary cultured neurons. J Proteome Res 6:2435–2446
Gianola S, Rossi F (2004) GAP-43 overexpression in adult mouse Purkinje cells overrides myelin-derived inhibition of neurite growth. Eur J Neurosci 19:819–830
Gnecchi M, He H, Melo LG, Noiseaux N, Morello F, de Boer RA, Zhang L, Pratt RE, Dzau VJ, Ingwall JS (2009) Early beneficial effects of bone marrow-derived mesenchymal stem cells overexpressing Akt on cardiac metabolism after myocardial infarction. Stem Cells 27:971–979 (Dayton, Ohio)
Grigoriadis N, Lourbopoulos A, Lagoudaki R, Frischer JM, Polyzoidou E, Touloumi O, Simeonidou C, Deretzi G, Kountouras J, Spandou E, Kotta K, Karkavelas G, Tascos N, Lassmann H (2011) Variable behavior and complications of autologous bone marrow mesenchymal stem cells transplanted in experimental autoimmune encephalomyelitis. Exp Neurol 230:78–89
Hofstetter CP, Schwarz EJ, Hess D, Widenfalk J, El Manira A, Prockop DJ, Olson L (2002) Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery. Proc Natl Acad Sci U S A 99:2199–2204
Isele NB, Lee HS, Landshamer S, Straube A, Padovan CS, Plesnila N, Culmsee C (2007) Bone marrow stromal cells mediate protection through stimulation of PI3-K/Akt and MAPK signaling in neurons. Neurochem Int 50:243–250
Kiedrowski L (1999) N-methyl-D-aspartate excitotoxicity: relationships among plasma membrane potential, Na(+)/Ca(2+) exchange, mitochondrial Ca(2+) overload, and cytoplasmic concentrations of Ca(2+), H(+), and K(+). Mol Pharmacol 56:619–632
Kilroy GE, Foster SJ, Wu X, Ruiz J, Sherwood S, Heifetz A, Ludlow JW, Stricker DM, Potiny S, Green P, Halvorsen YD, Cheatham B, Storms RW, Gimble JM (2007) Cytokine profile of human adipose-derived stem cells: expression of angiogenic, hematopoietic, and pro-inflammatory factors. J Cell Physiol 212:702–709
Kopen GC, Prockop DJ, Phinney DG (1999) Marrow stromal cells migrate through-out forebrain and cerebellum, and they differen tiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci U S A 96:10711–10716
Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch: Eur J Physiol 460:525–542
Li Y, Li H, Liu G, Liu Z (2013) Effects of neuregulin-1beta on growth-associated protein 43 expression in dorsal root ganglion neurons with excitotoxicity induced by glutamate in vitro. Neurosci Res 76:22–30
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622
Liu D, Pitta M, Mattson MP (2008) Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci 1147:275–282
Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP (2009) Nicotinamide prevents NAD+depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med 11:28–42
Liu Z, Cai H, Zhang P, Li H, Liu H, Li Z (2012) Activation of ERK1/2 and PI3K/Akt by IGF-1 on GAP-43 expression in DRG neurons with excitotoxicity induced by glutamate in vitro. Cell Mol Neurobiol 32:191–200
Liu R, Liu J, Ji X, Liu Y (2013) Synthetic nucleic acids delivered by exosomes: a potential therapeutic for generelated metabolic brain diseases. Metab Brain Dis 28:551–562
Lopatina T, Kalinina N, Karagyaur M, Stambolsky D, Rubina K, Revischin A, Pavlova G, Parfyonova Y, Tkachuk V (2011) Adipose-derived stem cells stimulate regeneration of peripheral nerves: BDNF secreted by these cells promotes nerve healing and axon growth de novo. PLoS One 6:e17899
Lu S, Lu C, Han Q, Li J, Du Z, Liao L, Zhao RC (2011) Adipose-derived mesenchymal stem cells protect PC12 cells from glutamate excitotoxicity-induced apoptosis by upregulation of XIAP through PI3-K/Akt activation. Toxicology 279:189–195
Martino G, Franklin RJ, Van Evercooren AB, Kerr DA (2010) Stem cell transplantation in multiple sclerosis: current status and future prospects. Nat Rev Neurol 6:247–255
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S (1999) Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann NY Acad Sci 893:154–175
McKinney RA, Luthi A, Bandtlow CE, Gahwiler BH, Thompson SM (1999) Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Natl Acad Sci U S A 96:11631–11636
Mochel F, Durant B, Meng X, O’Callaghan J, Yu H, Brouillet E, Wheeler VC, Humbert S, Schiffmann R, Durr A (2012) Early alterations of brain cellular energy homeostasis in Huntington disease models. J Biol Chem 287:1361–1370
Nicholls DG (2004) Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med 4:149–177
Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N (2007) Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res 85:3206–3212
Ohtaki H, Ylostalo JH, Foraker JE, Robinson AP, Reger RL, Shioda S, Prockop DJ (2008) Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc Natl Acad Sci U S A 105:14638–14643
Orrenius S (2004) Mitochondrial regulation of apoptotic cell death. Toxicol Lett 149:19–23
Parihar MS, Kunz EA, Brewer GJ (2008) Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res 86:2339–2352
Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair—current views. Stem Cells 25:2896–2902 (Dayton, Ohio)
Prockop DJ (2007) “Stemness” does not explain the repair of many tissues by mesenchymal srem/multipotent stromal cells (MSCs). Clin Pharmacol Ther 82:241–243
Raichle ME, Gusnard DA (2002) Appraising the brain’s energy budget. Proc Natl Acad Sci U S A 99:10237–10239
Salgado AJ, Reis RL, Sousa NJ, Gimble JM (2009) Adipose tissue derived stem cells secretome: soluble factors and their roles in regenerative medicine. Curr Stem Cell Res Ther 5:103–110
Sanchez-Ramos JR (2002) Neural cells derived from adult bone marr ow and umbilical cord blood. J Neurosci Res 69:880–893
Sheline CT, Behrens MM, Choi DW (2000) Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci: Off J Soc Neurosci 20:3139–3146
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI, Shulman RG, Rothman DL (1999) Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci USA 96:8235–8240
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998) Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci U S A 95:316–321
Singer NG, Caplan AI (2011) Mesenchymal stem cells: mechanisms of inflammation. Ann Rev Pathol 6:457–478
Skalnikova H, Motlik J, Gadher SJ, Kovarova H (2011) Mapping of the secretome of primary isolates of mammalian cells, stem cells and derived cell lines. Proteomics 11:691–708
Smaili SS, Ureshino RP, Rodrigues L, Rocha KK, Carvalho JT, Oseki KT, Bincoletto C, Lopes GS, Hirata H (2011) The role of mitochondrial function in glutamate-dependent metabolism in neuronal cells. Curr Pharm Des 17:3865–3877
Soane L, Kahraman S, Kristian T, Fiskum G (2007) Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J Neurosci Res 85:3407–3415
Uccelli A, Moretta L, Pistoia V (2008) Mesenchymal stem cells in health and disease. Nat Rev Immunol 8:726–736
Voulgari-Kokota A, Fairless R, Karamita M, Kyrargyri V, Tseveleki V, Evangelidou M, Delorme B, Charbord P, Diem R, Probert L (2012) Mesenchymal stem cells protect CNS neurons against glutamate excitotoxicity by inhibiting glutamate receptor expression and function. Exp Neurol 236:161–170
Zhang J, Li Y, Lu M, Cui Y, Chen J, Noffsinger L, Elias SB, Chopp M (2006) Bone marrow stromal cells reduce axonal loss in experimental autoimmune encephalomyelitis mice. J Neurosci Res 84:587–595
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295
Acknowledgments
This work was funded by the Chinese National Natural Science Foundation (No. 81071009,81271412, 31200740), the International S&T Cooperation Project of the Ministry of S&T of China (No. 2010DFR30850), the People’s Livelihood S&T Project, the Bureau of S&T of Dalian (No. 2010E11SF008, 2011E12SF030), and the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hao, P., Liang, Z., Piao, H. et al. Conditioned medium of human adipose-derived mesenchymal stem cells mediates protection in neurons following glutamate excitotoxicity by regulating energy metabolism and GAP-43 expression. Metab Brain Dis 29, 193–205 (2014). https://doi.org/10.1007/s11011-014-9490-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-014-9490-y