
Abstract
Immunophilins are a family of proteins encompassing FK506-binding proteins (FKBPs) and cyclophilins (Cyps). FKBPs and Cyps exert peptidyl-prolyl cis-trans isomerase (PPIase) activity, which facilitates diverse protein folding assembly, or disassembly. In addition, they bind to immunosuppressant medications where FKBPs bind to tacrolimus (FK506) and rapamycin, whereas cyclophilins bind to cyclosporin. Some large immunophilins have domains other than PPIase referred to as tetratricopeptide (TPR) domain, which is involved in heat shock protein 90 (Hsp90) and heat shock protein 70 (Hsp 70) chaperone interaction. The TPR domain confers immunophilins’ pleotropic actions to mediate various physiological and biochemical processes. So far, immunophilins have been implicated to play an important role in pathophysiology of inflammation, cancer and neurodegenerative disorders. However, their importance in the development of fibrosis has not yet been elucidated. In this review we focus on the pivotal functional and mechanistic roles of different immunophilins in fibrosis establishment affecting various organs. The vast majority of the studies reported that cyclophilin A, FKBP12 and FKBP10 likely induce organ fibrosis through the calcineurin or TGF-β pathways. FKBP51 demonstrated a role in myelofibrosis development through calcineurin-dependant pathway, STAT5 or NF-κB pathways. Inhibition of these specific immunophilins has been shown to decrease the extent of fibrosis suggesting that immunophilins could be a novel promising therapeutic target to prevent or reverse fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunophilins are a family of proteins that include FK506-binding proteins (FKBPs) and cyclophilins (Cyps) [1]. Both FKBPs and Cyps exert peptidyl-prolyl cis-trans isomerase (PPIase) activity, which catalyses the isomerization of proline cis-trans peptide bond [2], enhancing diverse protein folding assembly or disassembly. Irregular PPIase activity is associated with the development of cardiovascular disease, atherosclerosis, chronic kidney disease and type II diabetes [1]. In addition, they bind to immunosuppressant medications including tacrolimus (FK506) and rapamycin which bind to FKBPs and cyclosporin that binds to cyclophilins [3].
Notably, some large immunophilins have domains other than PPIase referred to as tetratricopeptide (TPR) domain, with main function involving chaperone interaction with heat shock protein 90 (Hsp90) and heat shock protein 70 (Hsp 70) [4]. These immunophilin-Hsp90/Hsp70 complexes facilitate secondary protein structure folding or unfolding, which is important for cell growth and differentiation [5]. The presence of additional domains confers immunophilins’ pleotropic function once bound to their respective target proteins such as glucocorticoid receptor and nuclear factor κB (NF-κB) to mediate various physiological and biochemical processes, including protein trafficking, receptor signalling, RNA recognition and transcription [6].
Based on their cellular distribution, immunophilins are classified as nuclear (e.g. FKBP25, Cyp33, FKBP13 and FKBPL) [7,8,9], mitochondrial (e.g. FKBP38, CypD) [10, 11], endoplasmic reticulum (e.g. CypB, FKBP13, FKBP19, FKBP22, FKBP60 and FKBP65) [12], cytoplasmic (e.g. FKBP12, CypA, and FKBPL) [13] and multidomain (e.g. FKBP36, FKBP38, FKBP51, FKBP52, and Cyp40) [14]. FKBP51 and CypA are mitochondrial factors that undergo nuclear-mitochondrial shuttling during stress response to protect cells against oxidative stress [15, 16]. The latter effect was also observed during cells differentiation, where FKBP51and CypA also translocate to the nucleus suggesting that they have a regulatory role in cells differentiation [17, 18]. Moreover, immunophilins are varied in their molecular weight, the higher the molecular weight the more complex the structure that lacks the immunosuppressant effect [19]. Parvulins belong to another group of proteins with PPIase activity outside of the immunophilin family, because they do not bind any specific immunosuppressive drug, hence not affecting protein folding and overall protein function [20]. FKBP like (FKBPL) is a divergent member of the immunophilin family that lacks the PPIase activity despite the presence of the PPI domain, therefore it is unable to bind to immunosuppressant drugs; however, it forms a complex with Hsp90 regulating downstream signalling [6].
In addition to the immunosuppressant effects, immunophilins have shown to have important roles in inflammation [21], cancer [22], cardiovascular disease and neurodegenerative disorders [23,24,25], by regulating soluble protein retrotransport through the interaction with dynein motors [4], neurodifferentiation and neuroregeneration [26], adipocytes differentiation [27], transcriptional regulation [28], steroid binding capacity [29], cell division [30] and apoptosis [31]. Even though fibrosis develops as a sequela of inflammation, the role of immunophilins in the process of fibrosis development has not yet been elucidated.
Fibrosis is defined as a process of excessive fibrous connective tissue accumulation containing collagen and fibronectin components of extracellular matrix (ECM), known to lead to organ dysfunction and subsequently depending on the organ in question heart failure, kidney disease, end-stage liver disease and idiopathic pulmonary fibrosis [32, 33]. Despite having distinct clinical manifestations, fibrosis is a result of chronic inflammation [34] induced by distinct triggering factors including recurrent exposure to smoke, irritants or toxins, myocardial infarction, obesity, elevated serum cholesterol and poorly controlled hypertension or diabetes [35]. However, regardless of the triggering factors, all fibrosis-associated disorders are characterised by the activation of ECM myofibroblasts towards tissue remodelling following tissue injury or damage [36]. For example, hepatitis C infection leads to myofibroblasts activation increasing collagen accumulation which distorts hepatic architecture, thereby, causing hepatocellular dysfunction and limiting hepatic blood flow causing portal hypertension [37]. On the other hand, left ventricular hypertrophy is associated with fibroblasts differentiation to myofibroblasts with increased synthesis of collagen and fibronectin that leads to ventricular dysfunction [38]. This extensive fibrosis and remodelling can ultimately lead to organ failure and death.
Notably, fibroblasts respond to paracrine signalling from macrophages and lymphocytes, as well as autocrine signalling to migration and differentiation to myofibroblasts [39] and increased secretion of growth factors, cytokines, and metalloproteinases (MMPs), and deposition of ECM proteins, thereby promoting fibrosis [40]. Multiple studies have revealed that TGFβ transcription with subsequent increase in TGFβ protein expression and downstream Smad signalling drives fibroblasts proliferation and differentiation [41] since TGFβ specifically induces α-smooth muscle actin (SMA) expression followed by collagen production [42], suggesting that both TGF-β and α-SMA play a key role in fibrosis pathogenesis. Meanwhile, renin-angiotensin aldosterone system (RAAS), cytokines (TNF-α, IL-21, TGF- β) [43], chemokines (MCP-1), angiogenic factors (VEGF) and caspases also appear to be dysregulated in fibrosis [44]. Therefore, they have been investigated as potential therapeutic targets of anti-fibrotic drugs.
Related to the role of immunophilins, FK506-binding proteins 10 (FKBP 10) was shown to interact with collagen [45], hence playing a crucial role in tissue remodelling [46]. This is suggested to occur through the peptidyl-prolyl cis-trans isomerase (PPIase) activity which is needed for proline isomerization facilitating collagen formation and assembly. Previous study has revealed that Fkbp10−/− mouse embryos display a low collagen crosslinking in calvarial collagen [47]. Furthermore, fibrosis due to TGF upregulation appears to be promoted by overexpression of FKBP51 [48]. Thus, this review outlines the key roles and mechanisms that various immunophilins play in fibrosis and discusses their therapeutic target potential towards the development of immunophilin inhibitors that could prevent fibrosis initiation and progression.
Key immunophilins in lung fibrosis development
Lung fibrosis is a progressive disease leading to scaring and stiffening of the lungs which eventually leads to respiratory failure [49]. In most cases, the diagnosis will be idiopathic pulmonary fibrosis; however, pulmonary fibrosis can be secondary to other causes, such as medications, radiation, environmental pollutants, infections, and genetic susceptibility [50, 51]. Although the pathogenic processes of pulmonary fibrosis are not completely understood [52], several studies have shown immunophilins to play an essential role.
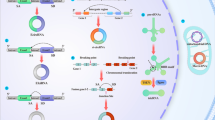
Cyclophilins are a family of proteins that facilitate protein folding and play a key role in fibrotic processes including inflammation, activation of apoptotic pathways, and activation of fibroblasts leading to increased collagen secretion [53]. Cyclophilins were found to be highly abundant in fibrotic tissues of the liver and mouth, and inhibition of cyclophilins by cyclosporin was reported to suppress the activity of calcineurin pathway, an important mechanism in fibrosis [54, 55]. Calcineurin (CaN) belongs to a superfamily of protein serine/threonine phosphatases and its activity is regulated by calcium/calmodulin. Following interaction between T-cell receptors with their ligands, calmodulin is activated due to the elevation of and interaction with the intracellular calcium level, activating its phosphatase activity and subsequently nuclear factor-activated T cells (NFAT) family members. NFAT then translocate into the nucleus and activates gene expression of cytokines including IL-2, IL-4, and CD40L which contribute to ECM remodelling, activation of collagen producing fibroblasts, and ultimately fibrosis (Fig. 1) [56]. Thus, inhibition of CaN/NFAT signalling pathway can prevent T-cell activation [57]. Additionally, basic fibroblast growth factor (bFGF) activates several signal factors that stimulate an increase in intracellular calcium levels, essential for cell transition from the G1 phase to the S phase that promote fibroblast proliferation and collagen synthesis with an important role in fibrosis [58, 59]. Yahong et al. [60] demonstrated that treating lung fibroblasts with bFGF increased its proliferation by two fold, in addition to 74% and 1.6-fold increase in collagen synthesis and secretion, respectively, which was associated with 60% increase in calcineurin activity. Interestingly, Cyclosporin A (CsA), calcineurin inhibitor, inhibited bFGF-stimulated lung fibroblasts proliferation by 66%, in addition to 37% and 56% inhibition in collagen synthesis and secretion, respectively, which was associated with 44% inhibition of calcineurin activity. Cyclosporin A exerts its effect through the inhibition of cyclophilin A, immunophilin protein member; this step prevents the phosphorylation of NFAT and its translocation into the nucleus therefore inhibiting T-cell activation [61], suggesting that cyclophilin A likely has an important role in lung fibroblasts activation and collagen secretion through CaN/NFAT pathway. Moreover, tacrolimus (FK506) is a calcineurin inhibitor used as an immunosuppressant agent for organ transplant rejection prevention, which exerts its effect through the inhibition of T lymphocytes by forming a complex with FKBP12 [62]. FKBP12 was found to interact with the extracellular domain TGF-beta receptor 1 (TβR-1) which is responsible for the initiation of the downstream signalling [63]. In vitro stimulation of the human lung fibroblasts cell line (TIG-20 cells) with TGF-β significantly increased collagen synthesis; however, treatment with tacrolimus prevented this increase in collagen synthesis. In line with the previous results, tacrolimus reduced the expression of TβR-1 in bleomycin-induced pulmonary fibrosis mice model, which suggests that FKBP12 could induce lung fibrosis through activation of TβR-1 [64]. Furthermore, Staab-Weijnitz et al. [46] demonstrated that FK506-binding protein 10 (FKBP10), another member of immunophilin family, is upregulated in lung protein lysates from bleomycin-induced lung fibrosis mouse model that was also confirmed using the microarray analysis of 99 lung samples from idiopathic pulmonary fibrosis patients showing an upregulation of FKBP10 gene expression compared to control, and this upregulation was positively correlated with α-SMA levels, a myofibroblasts marker. Interestingly, FKBP10 knockdown in idiopathic pulmonary fibrosis significantly reduced the expression of collagen I, V, and fibronectin. Since increased fibroblasts migration is a characteristic of idiopathic pulmonary fibrosis, KnÜppel et al. [65] studied the effect of FKBP10 deficiency on primary human lung fibroblast cell migration and adhesion. Following exposure to TGF-β1, the results showed that FKBP10 knockdown abrogated primary human lung fibroblast cell migration and adhesion, due to the reduction of collagen VI biosynthesis.

Cyclophilin A and FKBP12 activate T cells which induce fibrosis. Calcineurin (CaN) activity is regulated by Ca + 2/calmodulin. After the engagement of T-cell receptor with its ligand, intracellular Ca + 2 level will interact with calcineurin (CaN) and activates nuclear factor-activated T-cells (NFAT) family members. NFAT then translocate into the nucleus and activate gene expression of cytokines including IL-2, IL-4, and CD40L which then activate fibroblasts differentiation to myofibroblasts which induce extracellular matrix (ECM) synthesis and remodelling to induce fibrosis. Inhibition of CaN pathway with cyclosporin A (CsA) or FK506 will supress fibrosis
FKBP13, another member of immunophilin protein family, was reported to be highly expressed in lung biopsy samples from patients with idiopathic pulmonary fibrosis compared to control, which was also positively correlated with patient-reported dyspnoea scores. In addition, FKBP13 was positively correlated with α-SMA and unfolded protein response markers, GRP78 and total XBP1, expression, suggesting that higher levels of FKBP13 are associated with increased clinical severity and idiopathic pulmonary fibrosis pathogenicity [66]. In contrary, the same study revealed that FKBP13 knockout mice had a higher sensitivity to damaging effects of bleomycin through contributing to increased inflammatory cells infiltration, lung elastance and fibrosis and impaired resolution of fibrosis, therefore suggesting that FKBP13 might have a protective role against bleomycin-induced lung damage.
Taken together, cyclophilin A, FKBP12, FKBP10, and, FKBP13 have shown an important role in the pathogenesis of lung fibrosis; however, further research should be conducted to strengthen their therapeutic target potential in lung fibrosis treatment.
Immunophilins and liver fibrosis
Hepatic injury due to hepatitis B (HBV), hepatitis C (HCV), non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH), the severe form of NAFLD, often leads to hepatic fibrosis and subsequently advanced liver disease [67]. Persistent inflammation and activation of hepatic stellate cells (HSCs) are some of the main characteristics of hepatic fibrosis that lead to tissue remodelling and repair through accumulation of collagen. Progression of liver fibrosis to liver cirrhosis is associated with poor survival and hepatocellular carcinoma development [68]. Therefore, understanding the mechanisms underpinning the development of liver fibrosis is key and can lead to identification of new therapeutic targets.
Nakamuta et al. 2005 [69] studied the effect of cyclosporin A, a cyclophilins inhibitor, on HSCs growth and collagen production. In this study, cyclosporin A inhibited cell growth and collagen production through inhibition of c-jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPKs) phosphorylation (Fig. 2). Similarly, NIM881, cyclosporin A analogue, reduced HSCs growth, collagen production, in addition to increasing collagenase activity and phosphorylation of JNK and p38 [70]. In another study, a novel cyclophilin A inhibitor, NV556, was evaluated for its anti-fibrotic properties using two animal models, Methionine–Cholin-Deficient (MCD) diet model, and STAM model of Nonalcoholic Steatohepatitis model, as well as 3D human liver scaffold in vitro model. NV556 significantly reduced collagen deposition measured by percentage of Sirius red-positive area in the liver in the two animal models and reduced collagen IV expression in addition to significant reduction in LOX gene expression, a marker of activated hepatic stellate cells in vitro [53]. Thus, cyclophilin A inhibitors show promise as future treatments for liver fibrosis through inhibition of the JNK and p38 pathways. Moreover, it is well-established that human immunodeficiency virus-1 (HIV-1) co-infection with hepatitis C virus (HCV) increases the risk of liver fibrosis development [71]. In order to assess the effect of another cyclophilin A inhibitor, CPI-431-32, Gallay et al. developed a novel in vitro HCV and HIV-1 co-infection model, including human hepatocytes and CD4 + T lymphocytes. The results of this study demonstrated that CPI-431-32 is capable of inhibiting the replication of both HCV and HIV-1 and their variants [72]. Which suggests that cyclophilin A inhibition during HCV infection could prevent viral replication and ultimately liver fibrosis. Another important mechanism implicated in HCV infection which involves FKBP38, another member of immunophilins. FKBP38 binds to and inhibits mammalian target of rapamycin (mTOR), and consequently, inactivated mTOR fails to phosphorylate downstream targets S6K1 and 4EBP1, which promote cell apoptosis. When cells are infected with HCV, it competes with mTOR for interacting with FKBP38, resulting in the dissociation of mTOR from FKBP38. Thus, activated mTOR phosphorylates downstream targets S6K1 and 4EBP1, which suppress cell apoptosis [73]. Therefore, FKBP38 is essential for HCV persistence and the development of HCV-induced liver fibrosis [74].

Cyclophilin A activates JNK and MAPK-P38 which induce liver fibrosis. Cyclophilin A activates phosphorylation of c-jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPKs) which induce hepatic stellate cells (HSCs) growth and collagen production that induce liver fibrosis. Cyclosporin A inhibited cell growth and collagen production which was associated with suppressed phosphorylation of c-jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPKs)
Important roles of immunophilins in cardiac fibrosis and heart disease
Cardiac fibrosis is integral component of many different forms of heart disease. Since the regenerative capacity of the mammalian myocardium is limited, sudden loss of a large number of cardiomyocytes initiates an inflammatory response that replaces the dead myocardium with collagen-based scar [75, 76]. Although, a number of different pathophysiologic conditions can induce cardiac fibrosis including myocardial infarction, ageing, pressure overload, volume overload, hypertrophic cardiomyopathy, diabetes, obesity and toxic insults, the cellular pathogenic mechanism are often similar [77,78,79,80].
As discussed above, calcineurin is a calcium-dependent phosphatase that dephosphorylates NFATs which then translocates to the nucleus and activates inflammatory response genes. NFATs are expressed highly in T cells and skeletal muscles, whereas NFAT3 is expressed in different tissues, including the heart. Activation of calcineurin dephosphorylates NFAT3 inducing nuclear expression of cytokines that activate T cells (Fig. 1) [81]. Molkentin et al. demonstrated that calcineurin transgenic mice were highly susceptible to sudden death partially due to fibrosis of the ventricular wall. Similar results were demonstrated in NFAT3 transgenic mice showing an extensive fibrosis in the cardiac ventricular wall. Interestingly, as with liver and lung fibrosis, a cyclophilin A inhibitor prevented cardiac fibrosis which was demonstrated in calcineurin transgenic mice [82]. Another immunophilin inhibitor, FK506/FKBP12 inhibitor, was capable of attenuating angiotensin II (Ang II)-induced increase in ERK1/2 and p38 MAPK phosphorylation using rat cardiac fibroblasts. In addition, Ang II-induced rat cardiac fibroblasts proliferation in conjunction with upregulation of fibronectin, pro-collagen, inducible nitric oxide (iNOS) and inflammatory cytokines were inhibited by both cyclophilin A and FK506 inhibitors, suggesting that these inhibitors could attenuate cardiac fibrosis trough inhibition of the calcineurin pathway [83]. In line with the in vitro results in the previous study, inhibition of cyclophilin A and FKBP12 reduced the extent of cardiac fibrosis by inhibition of calcineurin activation in load-induced cardiac hypertrophy rat model, providing further evidence for an important role of cyclophilin A and FKBP12 in inducing cardiac fibrosis through calcineurin pathway [84, 85]. Consistent with the previous reports, cyclophilin A expression is increased in a mouse model of troponin I-induced autoimmune myocarditis, associated with severe cardiac fibrosis, and the inhibition of cyclophilin A with MM284 markedly reduced cardiac fibrosis. When monocytes migration and adhesion was stimulated with recombinant cyclophilin A, there was a significant increase in TNF-α, IL-6, and MMP-9 expression, which was abrogated following addition of cyclophilin A inhibitor, MM284, treatment [86]. Satoh et al. showed a marked decreased in collagen content in the perivascular area in apolipoprotein E and cyclophilin A knockout mice model (Apoe−/− Ppia−/−) after angiotensin II treatment. These results were evaluated also by isolating cardiac fibroblasts from Apoe−/− and Apoe−/− Ppia−/− mice and determining fibroblasts proliferation and production of reactive oxygen species (ROS) after Ang II treatment. Whilst, cardiac fibroblast proliferation and ROS production were increased from Apoe−/− mice, a dramatic reduction in ROS production was observed in Apoe−/− Ppia−/− mice with no change in proliferation. Additionally, the growth rate of cardiac fibroblasts was higher in Apoe−/− compared to Apoe−/− Ppia−/− fibroblasts and treating cardiac fibroblasts with recombinant cyclophilin A increased cardiac fibroblasts proliferation and migration, which suggest that cyclophilin A contributes to cardiac fibrosis [87]. In a rat-reduced uterine perfusion pressure model of preeclampsia, cardiovascular disorder of pregnancy, cardiac fibrosis was observed measured by the extent of collagen deposition in the heart, and this was in association with a significant increase in cardiac mRNA and protein expression of FK506-binding protein like (FKBPL), a divergent member of the immunophilin family and a key angiogenesis-related protein [24, 88, 89]. Aligned work also demonstrated an increase in FKBPL expression in human cardiac fibroblast cell line exposed to fibrotic stimuli, TGF-β [90]. In a separate study using human plasma samples, high systemic FKBPL levels were reported in people with cardiovascular disease including diastolic dysfunction and established preeclampsia [89, 91]. Whilst the role of FKBPL in cardiac fibrosis is still not fully elucidated, future research should address its potential as both therapeutic target and a biomarker of cardiac fibrosis to enable early diagnosis and prevention of subsequent heart disease.
FKBP51 importance in myelofibrosis development
Idiopathic myelofibrosis is a myeloproliferative disease characterised by clonal stem cell dysfunction that leads to megakaryocyte hyperplasia and fibrotic cytokines release within the bone marrow environment [92]. Ineffective haematopoiesis leading to pancytopenia and extramedullary haematopoiesis are the main symptoms for this disease due to the collagen deposition in bone marrow tissue [93].
FKBP51 is a member of immunophilin family that can regulate FK506-induced calcineurin inhibition and it was found to be overexpressed in megakaryocytes derived from idiopathic myelofibrosis patients compared to normal megakaryocytes [94]. Furthermore, overexpression of FKBP51 in human megakaryoblastic leukaemia cells, UT-7/mpl, markedly inhibited calcineurin activity, which was associated with induced resistance to apoptosis mediated by cytokines deprivation, suggesting that FKBP51 could be responsible for megakaryocytes hyperplasia through calcineurin-dependant pathway. Moreover, Komura et al. demonstrated that in FKBP51-overexpresssing cell line, STAT5 was sustainably activated in association with JAK2 phosphorylation implying the importance of this mechanism for spontaneous growth of megakaryocytes in idiopathic myelofibrosis In addition, in 2005, Komura et al. also showed that FKBP51 overexpression in UT-7/mpl cell line induced a sustained activation for the nuclear factor κB (NF-κB) after cytokine deprivation and this activation of NF-κB was also detected in proliferating megakaryocytes and in circulating CD34 + patient cells [48]. Interestingly, the inhibition of NF-κB activity did not alter the apoptotic resistance of UT-7/mpl cells and CD34 + megakaryocytes derived from idiopathic myelofibrosis patients, but, inhibited TGF-β1 secretion, highlighting the importance of NF-κB activation in the development of fibrosis in this disease. Taken together, FKBP51 overexpression in idiopathic myelofibrosis disease cells could play an important role in the pathogenesis of this disease.
Future implications
Overall, a number of immunophilins including cyclophilin A, FKBP12, FKBP38, FKBP51 and FKBPL have shown emerging roles as important pathogenic mechanisms in the development of fibrosis in different organs. However, further research is needed to fully elucidate the therapeutic target or biomarker potential of these immunophilins in lung, liver, myelofibrosis and cardiac fibrosis towards clinical translation and development of much needed anti-fibrotic agents. Furthermore, the importance of immunophilins in fibrosis of other organ including kidneys and gastrointestinal tract should be explored. Although, the vast majority of the studies assessed inhibitors of the conventional immunophilin-based immunosuppressants, cyclosporin A and tacrolimus, these therapeutic strategies are hindered by a number of dangerous side effects related to the immune system. Tacrolimus was able to activate TGF-β signalling in endothelial cells which caused renal arteriolar hyalinosis in renal transplant patients [96]. In addition, immunophilin-based immunosuppressants can cause chronic allograft vasculopathy associated with endothelial oxidative stress, apoptosis and dysfunction that affect the half-life-engrafted solid organ negatively [97]. Moreover, tacrolimus and cyclosporin are able to induce toll-like receptor-4 (TLR4) and the downstream NF-κB that lead to the activation of endothelial cells and increase the production of pro-inflammatory mediators [98]. Therefore, the development of novel therapeutic agents that target other immunophilins, particularly FKBPs, potentially with better side effect profile could be a more viable approach to preventing or reversing fibrosis.
Summary and conclusion
In this review, we discussed the importance of various immunophilin proteins in organ fibrosis and their downstream signalling that could contribute to the pathogenesis of fibrosis, Table 1. Given this is an area that is still poorly understood with limited research conducted so far, the focus has been on the processes of lung, liver, cardiac fibrosis and myelofibrosis in bone marrow development, which is an area of unmet clinical need with ineffective therapeutic options. The vast majority of scientific reports investigated the importance of targeting cyclophilin A in lung, liver and cardiac fibrosis, showing that this therapeutic strategy can reduce organ fibrosis through inhibition of the CaN/NFAT pathway. FKBP12 appears to have some contribution to the pathogenesis of lung and cardiac fibrosis through the TGF-beta and calcineurin pathway. Similarly, a few studies showed that FKBP10 is implicated in inducing lung fibrosis through the activation of the TGF-beta pathway, whereas FKBP13 showed a conflicting role in lung fibrosis. FKBPL is emerging as a potential biomarker and therapeutic target of cardiac fibrosis; however, this needs to be elucidated further. FKBP51 demonstrated a role in myelofibrosis development through calcineurin-dependent pathway, STAT5 or NF-κB pathways. In conclusion, some members of immunophilin protein family have shown a promising role in organ fibrosis development; however, this is an under-research area that needs further evidence in order to progress immunophilin-based therapeutic or biomarker strategies towards clinical utilisation.
Data availability
Not applicable.
References
McClements L, Annett S, Yakkundi A, Robson T (2015) The role of peptidyl prolyl isomerases in aging and vascular diseases. Curr Mol Pharmacol 9:165–179. https://doi.org/10.2174/1874467208666150519115729
Dugave C (2005) Study of the cis-trans isomerization of the amino-acyl prolyl peptide bond application to the design of novel inhibitors of immunophilins. Curr Org Chem. https://doi.org/10.2174/1385272023373301
Ghartey-Kwansah G, Li Z, Feng R, Wang L, Zhou X, Chen FZ, Meng Meng Xu et al (2018) Comparative analysis of FKBP family protein: evaluation, structure, and function in mammals and Drosophila melanogaster. BMC Dev Biol. https://doi.org/10.1186/s12861-018-0167-3
Zgajnar NR, De Leo SA, Lotufo CM, Erlejman AG, Pilipuk GP, Galigniana MD (2019) Biological actions of the hsp90-binding immunophilins FKBP51 and FKBP52. Biomolecules 9:1–30. https://doi.org/10.3390/biom9020052
Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB (2001) Evidence that the peptidylprolyl isomerase domain of the hsp90-binding immunophilin FKBP52 Is involved in both dynein interaction and glucocorticoid receptor movement to the nucleus. J Biol Chem 276:14884–14889. https://doi.org/10.1074/JBC.M010809200
Annett S, Moore G, Robson T (2020) FK506 binding proteins and inflammation related signalling pathways; basic biology, current status and future prospects for pharmacological intervention. Pharmacol Ther 215:107623. https://doi.org/10.1016/j.pharmthera.2020.107623
Rajiv C, Davis TL (2018) Structural and functional insights into human nuclear cyclophilins. Biomolecules 8:161. https://doi.org/10.3390/BIOM8040161
Robson T, James IF (2012) The therapeutic and diagnostic potential of FKBPL: a novel anticancer protein. Drug Discov Today. https://doi.org/10.1016/j.drudis.2012.01.002
Prakash A, Shin J, Yoon HS (2015) (1)H, (13)C and (15)N resonance assignments of human FK506 binding protein 25. Biomol NMR Assign 9:43–46. https://doi.org/10.1007/S12104-014-9541-7
Amanakis G, Murphy E (2020) Cyclophilin D: an integrator of mitochondrial function. Front Physiol 11:595. https://doi.org/10.3389/FPHYS.2020.00595/XML/NLM
Lu Q, Wang M, Gui Y, Hou Q, Mengru G, Liang Y, Xiao B, Zhao AZ, Dai C (2020) Rheb1 protects against cisplatin-induced tubular cell death and acute kidney injury via maintaining mitochondrial homeostasis. Cell Death Dis. https://doi.org/10.1038/s41419-020-2539-4
Kolos JM, Voll AM, Bauder M, Hausch F (2018) FKBP ligands—where we are and where to go? Front Pharmacol 9:1425. https://doi.org/10.3389/FPHAR.2018.01425/XML/NLM
Xing M, Wang J, Qin Yang Y, Wang JL, Xiong J, Zhou S (2019) FKBP12 is a predictive biomarker for efficacy of anthracycline-based chemotherapy in breast cancer. Cancer Chemother Pharmacol 84:861–872. https://doi.org/10.1007/S00280-019-03923-1/FIGURES/4
Harikishore A, Yoon HS (2015) Immunophilins: structures, mechanisms and ligands. Curr Mol Pharmacol 9:37–47. https://doi.org/10.2174/1874467208666150519113427
Gallo LI, Lagadari M, Piwien-Pilipuk G, Galigniana MD (2011) The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress. J Biol Chem 286:30152–30160. https://doi.org/10.1074/jbc.M111.256610
Daneri-Becerra C, Valeiras B, Gallo LI, Lagadari M, Galigniana MD (2021) Cyclophilin A is a mitochondrial factor that forms complexes with p23 - correlative evidence for an anti-apoptotic action. J Cell Sci. https://doi.org/10.1242/jcs.253401
Toneatto J, Guber S, Charó NL, Susperreguy S, Schwartz J, Galigniana MD, Piwien-Pilipuk G (2013) Dynamic mitochondrial-nuclear redistribution of the immunophilin FKBP51 is regulated by the PKA signaling pathway to control gene expression during adipocyte differentiation. J Cell Sci 126:5357–5368. https://doi.org/10.1242/JCS.125799/263521/AM/DYNAMIC-MITOCHONDRIAL-NUCLEAR-REDISTRIBUTION-OF
Chiu R, Rey O, Zheng JQ, Twiss JL, Song J, Pang S, Yokoyama KK (2003) Effects of altered expression and localization of Cyclophilin A on differentiation of p19 embryonic carcinoma cells. Cell Mol Neurobiol 23:929–943. https://doi.org/10.1023/B:CEMN.0000005321.11544.cc
Wiederrecht G, Hung S, Chan HK, Marcy A, Martin M, Calaycay J, Boulton D, Sigal N, Kincaid RL, Siekierka JJ (1992) Characterization of high molecular weight FK-506 binding activities reveals a novel FK-506-binding protein as well as a protein complex. J Biol Chem 267:21753–21760. https://doi.org/10.1016/s0021-9258(19)36676-1
Olejnik P, Mądrzak CJ, Nuc K (2021) Cyclophilins and their functions in abiotic stress and plant–microbe interactions. Biomolecules. https://doi.org/10.3390/biom11091390
Stephanie A, Spence S, Garciarena C, Campbell C, Dennehy M, Drakeford C, Lai J et al (2021) The immunophilin protein FKBPL and its peptide derivatives are novel regulators of vascular integrity and inflammation via NF-κB signalling. bioRxiv. https://doi.org/10.1101/2021.02.24.431422
Leo De, Sonia A, Zgajnar NR, Mazaira GI, Erlejman AG, Galigniana MD (2019) Role of the Hsp90-immunophilin heterocomplex in cancer biology. Curr Cancer Ther Rev 16:19–28. https://doi.org/10.2174/1573394715666190102120801
McNally R, Alqudah A, McErlean EM, Rennie C, Morshed N, Short A, McGrath K et al (2021) Non-viral gene delivery utilizing RALA modulates sFlt-1 secretion, important for preeclampsia. Nanomedicine 16:1999–2012. https://doi.org/10.2217/NNM-2021-0180
Richards C, Sesperez K, Chhor M, Ghorbanpour S, Rennie C, Ming CLC, Evenhuis C et al (2021) Characterisation of cardiac health in the reduced uterine perfusion pressure model and a 3D cardiac spheroid model, of preeclampsia. Biol Sex Diff 12:1–14. https://doi.org/10.1186/s13293-021-00376-1
Blair LJ, Nordhues BA, Hill SE, Matthew Scaglione K, O’Leary JC, Fontaine SN, Breydo L et al (2013) Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J Clin Invest 123:4158–4169. https://doi.org/10.1172/JCI69003
Daneri-Becerra C, Galigniana M (2022) The Hsp90-binding immunophilin FKBP52 enhances neurodifferentiation and neuroregeneration in murine models. Neural Regen Res 17:555. https://doi.org/10.4103/1673-5374.320976
Häusl AS, Balsevich G, Gassen NC, Schmidt MV (2019) Focus on FKBP51: a molecular link between stress and metabolic disorders. Mol Metab 29:170–181. https://doi.org/10.1016/J.MOLMET.2019.09.003
Orłowski M, Popławska K, Pieprzyk J, Szczygieł-Sommer A, Wiȩch A, Zarȩbski M, Tarczewska A, Dobrucki J, Ozyhar A (2018) Molecular determinants of Drosophila immunophilin FKBP39 nuclear localization. Biol Chem 399:467–484. https://doi.org/10.1515/HSZ-2017-0251
Mazaira GI, Echeverría PC, Ciucci SM, Monte M, Gallo LI, Erlejman AG, Galigniana MD (2021) Differential regulation of the glucocorticoid receptor nucleocytoplasmic shuttling by TPR-domain proteins. Biochim et Biophys Acta Mol Cell Res 1868:119000. https://doi.org/10.1016/J.BBAMCR.2021.119000
Dilworth D, Gudavicius G, Xiaoxue X, Boyce AKJ, O’Sullivan C, Serpa JJ, Bilenky M et al (2018) The prolyl isomerase FKBP25 regulates microtubule polymerization impacting cell cycle progression and genomic stability. Nucleic Acids Res 46:2459–2478. https://doi.org/10.1093/NAR/GKY008
Ma Z, Zhang W, Yaru W, Zhang M, Wang L, Wang Y, Wang Y, Liu W (2021) Cyclophilin A inhibits A549 cell oxidative stress and apoptosis by modulating the PI3K/Akt/mTOR signaling pathway. Biosci Rep. https://doi.org/10.1042/BSR20203219/227464
Kisseleva T, Brenner D (2021) Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 18:151–166. https://doi.org/10.1038/s41575-020-00372-7
Maher TM, Strek ME (2019) Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir Res 20:1–9. https://doi.org/10.1186/S12931-019-1161-4/FIGURES/7
Tanwar S, Rhodes F, Srivastava A, Trembling PM, Rosenberg WM (2020) Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J Gastroenterol 26:109. https://doi.org/10.3748/WJG.V26.I2.109
Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R (2019) Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 15:705–730. https://doi.org/10.1038/s41584-019-0322-7
Moretti L, Stalfort J, Barker TH, Abebayehu D (2022) The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J Biol Chem 298:101530. https://doi.org/10.1016/J.JBC.2021.101530
Rajapaksha IG, Gunarathne LS, Angus PW, Herath CB (2021) Update on new aspects of the renin-angiotensin system in hepatic fibrosis and portal hypertension: implications for novel therapeutic options. J Clin Med 10:1–20. https://doi.org/10.3390/jcm10040702
Zhao W, Zhao J, Rong J (2020) Pharmacological modulation of cardiac remodeling after myocardial infarction. Oxid Med Cell Longev. https://doi.org/10.1155/2020/8815349
Tarbit E, Singh I, Peart JN, Rose’Meyer RB (2019) Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail Rev 24:1–15. https://doi.org/10.1007/S10741-018-9720-1/TABLES/2
Segers VFM, De Keulenaer GW (2021) Autocrine signaling in cardiac remodeling: a rich source of therapeutic targets. J Am Heart Assoc 10:1–28. https://doi.org/10.1161/JAHA.120.019169
Tang PM, Kuen YY, Zhang TS, Mak K, Tang PCT, Huang XR, Lan HY (2018) Transforming growth factor-β signalling in renal fibrosis: from Smads to non-coding RNAs. J Physiol 596:3493–3503. https://doi.org/10.1113/JP274492
Zhang HY, Gharaee-Kermani M, Zhang K, Karmiol S, Phan SH (1996) Lung fibroblast α-smooth muscle actin expression and contractile phenotype in bleomycin-induced pulmonary fibrosis. Am J Pathol. 148:527
AlQudah M, Hale TM, Czubryt MP (2020) Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol 91–92:92–108. https://doi.org/10.1016/j.matbio.2020.04.005
Balzer MS (2020) Molecular pathways in peritoneal fibrosis. Cell Signal 75:109778. https://doi.org/10.1016/j.cellsig.2020.109778
Ishikawa Y, Vranka J, Wirz J, Nagata K, Bächinger HP (2008) The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J Biol Chem. https://doi.org/10.1074/jbc.M802535200
Staab-Weijnitz CA, Fernandez IE, Knüppel L, Maul J, Heinzelmann K, Juan-Guardela BM, Hennen E et al (2015) FK506-binding protein 10, a potential novel drug target for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 192:455–467. https://doi.org/10.1164/RCCM.201412-2233OC/SUPPL_FILE/DISCLOSURES.PDF
Lietman CD, Rajagopal A, Homan EP, Munivez E, Jiang MM, Bertin TK, Chen Y et al (2014) Connective tissue alterations in Fkbp10-/- mice. Hum Mol Genet. https://doi.org/10.1093/hmg/ddu197
Komura E, Tonetti C, Penard-Lacronique V, Chagraoui H, Lacout C, LeCouédic JP, Rameau P, Debili N, Vainchenker W, Giraudier S (2005) Role for the nuclear factor κB pathway in transforming growth factor-β1 production in idiopathic myelofibrosis: possible relationship with FK506 binding protein 51 overexpression. Can Res 65:3281–3289. https://doi.org/10.1158/0008-5472.CAN-04-2339
Richeldi L, Collard HR, Jones MG (2017) Idiopathic pulmonary fibrosis. The Lancet 389:1941–1952. https://doi.org/10.1016/S0140-6736(17)30866-8
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU (2017) Idiopathic pulmonary fibrosis. Nat Rev Dis Primer 3:1–20. https://doi.org/10.1038/nrdp.2017.74
Mirjam K, Roldan N, Machahua C, Sengupta A, Geiser T, Guenat OT, Funke-Chambour M, Hobi N, Kruithof-de Julio M (2021) Human-based advanced in vitro approaches to investigate lung fibrosis and pulmonary effects of COVID-19. Front Med 8:555. https://doi.org/10.3389/FMED.2021.644678/BIBTEX
Martin MD, Chung JH, Kanne JP (2016) Idiopathic pulmonary fibrosis. J Thorac Imaging 31:127–139. https://doi.org/10.1097/RTI.0000000000000204
Serrano S, Sonia AG, Longato L, Rombouts K, Kuo J, Gregory M, Moss S et al (2019) Evaluation of NV556, a novel Cyclophilin inhibitor, as a potential antifibrotic compound for liver fibrosis. Cells 8:1–15. https://doi.org/10.3390/cells8111409
Hou X, Liu R, Canhua Huang Lu, Zhou JY, Chen Q (2017) Cyclophilin A was revealed as a candidate marker for human oral submucous fibrosis by proteomic analysis. Cancer Biomark 20:345–356. https://doi.org/10.3233/CBM-170142
Takeda Y, Yoneda T, Demura M, Usukura M, Mabuchi H (2002) Calcineurin inhibition attenuates mineralocorticoid-induced cardiac hypertrophy. Circulation 105:677–679. https://doi.org/10.1161/HC0602.104675
Bradshaw AD, DeLeon-Pennell KY (2020) T-cell regulation of fibroblasts and cardiac fibrosis. Matrix Biol 91–92:167–175. https://doi.org/10.1016/J.MATBIO.2020.04.001
Matsuda S, Koyasu S (2000) Mechanisms of action of cyclosporine. Immunopharmacology 47:119–125
Labram B, Namvar S, Hussell T, Herrick SE (2019) Endothelin-1 mediates Aspergillus fumigatus-induced airway inflammation and remodelling. Clin Exp Allergy 49:861–873. https://doi.org/10.1111/CEA.13367
Antunes S, Ricardo AK, Mehta LM, Tocker J, Croft M (2018) TNFSF14 (LIGHT) exhibits inflammatory activities in lung fibroblasts complementary to IL-13 and TGF-β. Front Immunol 9:576. https://doi.org/10.3389/FIMMU.2018.00576/BIBTEX
Yahong C, Mingwu Z, Mingui F, Wanzhen Y (2003) The role of calcineurin in the lung fibroblasts proliferation and collagen synthesis induced by fibroblasts growth factor. Chin Med J 345:857–862
Tedesco D, Haragsim L (2012) Cyclosporine: a review. J Transplant 2012:1–7. https://doi.org/10.1155/2012/230386
Fruman DA, Wood MA, Gjertson CK, Katz HR, Burakoff SJ, Bierer BE (1995) FK506 binding protein 12 mediates sensitivity to both FK506 and rapamycin in murine mast cells. Eur J Immunol 25:563–571. https://doi.org/10.1002/EJI.1830250239
Wang T, Donahoe PK (2004) The immunophilin FKBP12: a molecular guardian of the TGF-β family type I receptors. Front Biosci 9:619
Nagano J, Iyonaga K, Kawamura K, Yamashita A, Ichiyasu H, Okamoto T, Suga M, Sasaki Y, Kohrogi H (2006) Use of tacrolimus, a potent antifibrotic agent, in bleomycin-induced lung fibrosis. Eur Respir J. https://doi.org/10.1183/09031936.06.00070705
Knüppel L, Heinzelmann K, Lindner M, Hatz R, Behr J, Eickelberg O, Staab-Weijnitz CA (2018) FK506-binding protein 10 (FKBP10) regulates lung fibroblast migration via collagen VI synthesis. Respir Res. https://doi.org/10.1186/s12931-018-0768-1
Tat V, Ayaub EA, Ayoub A, Vierhout M, Naiel S, Padwal MK, Abed S et al (2021) Fk506-binding protein 13 expression is upregulated in interstitial lung disease and correlated with clinical severity a potentially protective role. Am J Respir Cell Mol Biol 64:235–246. https://doi.org/10.1165/RCMB.2020-0121OC
Bataller R, Rombouts K, Altamirano J, Marra F (2011) Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Practice Res Clin Gastroenterol 25:231–244. https://doi.org/10.1016/J.BPG.2011.02.010
Carloni V, Luong TV, Rombouts K (2014) Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int 34:834–843. https://doi.org/10.1111/LIV.12465
Nakamuta M, Kohjima M, Fukushima M, Morizono S, Kotoh K, Kobayashi N, Enjoji M (2005) Cyclosporine suppresses cell growth and collagen production in hepatic stellate cells. Transpl Proc 37:4598–4602. https://doi.org/10.1016/j.transproceed.2005.10.104
Kohjima M, Enjoji M, Higuchi N, Kotoh K, Kato M, Takayanagi R, Nakamuta M (2007) NIM811, a nonimmunosuppressive cyclosporine analogue, suppresses collagen production and enhances collagenase activity in hepatic stellate cells. Liver Int 27:1273–1281. https://doi.org/10.1111/j.1478-3231.2007.01560.x
Lin W, Weinberg EM, Chung RT (2013) Pathogenesis of accelerated fibrosis in HIV/HCV Co-infection. J Infect Dis 207:S13–S18. https://doi.org/10.1093/INFDIS/JIS926
Gallay PA, Bobardt MD, Chatterji U, Trepanier DJ, Ure D, Ordonez C, Foster R (2015) The novel cyclophilin inhibitor CPI-431-32 concurrently blocks HCV and HIV-1 infections via a similar mechanism of action. PLoS ONE 10:1–18. https://doi.org/10.1371/journal.pone.0134707
Shrivastava S, Chowdhury JB, Steele R, Ray R, Ray RB (2012) Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol 86:8705. https://doi.org/10.1128/JVI.00616-12
Peng L, Liang D, Tong W, Li J, Yuan Z (2010) Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J Biol Chem 285:20870–20881. https://doi.org/10.1074/jbc.M110.112045
Berk BC, Fujiwara K, Lehoux S (2007) ECM remodeling in hypertensive heart disease. J Clin Invest 117:568–575. https://doi.org/10.1172/JCI31044
Frangogiannis NG (2012) Regulation of the inflammatory response in cardiac repair. Circ Res 110:159–173. https://doi.org/10.1161/CIRCRESAHA.111.243162
Borer JS, Truter S, Herrold EM, Falcone DJ, Pena M, Carter JN, Dumlao TF, Lee JA, Supino PG (2002) Myocardial fibrosis in chronic aortic regurgitation molecular and cellular responses to volume overload. Circulation 105:1837–1842. https://doi.org/10.1161/01.CIR.0000014419.71706.85
Ashrafian H, McKenna WJ, Watkins H (2011) Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res 109:86–96. https://doi.org/10.1161/CIRCRESAHA.111.242974
Asbun J, Villarreal FJ, City M, Diego S (2006) The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am College Cardiol. https://doi.org/10.1016/j.jacc.2005.09.050
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71:549–574. https://doi.org/10.1007/S00018-013-1349-6
Hoey T, Sun Y-L, Williamson K, Xiang Xu (1995) Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 2:461–472
Molkentin JD, Jian Rong Lu, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228. https://doi.org/10.1016/S0092-8674(00)81573-1
White M, Montezano AC, Touyz RM (2012) Angiotensin II signalling and calcineurin in cardiac fibroblasts: differential effects of calcineurin inhibitors FK506 and cyclosporine A. Ther Adv Cardiovasc Dis 6:5–14. https://doi.org/10.1177/1753944711432901
Shimoyama M, Hayashi D, Takimoto E, Zou Y, Oka T, Uozumi H, Kudoh S et al (1999) Calcineurin plays a critical role in pressure overload-induced cardiac hypertrophy. Circulation 100:2449–2454. https://doi.org/10.1161/01.CIR.100.24.2449
Lim HW, De Windt LJ, Steinberg L, Taigen T, Witt SA, Kimball TR, Molkentin JD (2000) Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation 101:2431–2437
Heinzmann D, Bangert A, Müller AM, Von Ungern-Sternberg SNI, Emschermann F, Schönberger T, Chatterjee M et al (2015) The novel extracellular Cyclophilin A (CyPA) - Inhibitor MM284 reduces myocardial inflammation and remodeling in a mouse model of troponin I -induced myocarditis. PLoS ONE 10:1–11. https://doi.org/10.1371/journal.pone.0124606
Satoh K, Nigro P, Zeidan A, Soe NN, Jaffré F, Oikawa M, O’Dell MR et al (2011) Cyclophilin a promotes cardiac hypertrophy in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31:1116–1123. https://doi.org/10.1161/ATVBAHA.110.214601
Alqudah A, Eastwood KA, Jerotic D, Todd N, Hoch D, McNally R, Obradovic D et al (2021) FKBPL and SIRT-1 are downregulated by diabetes in pregnancy impacting on angiogenesis and endothelial function. Front Endocrinol. https://doi.org/10.3389/FENDO.2021.650328
Todd N, McNally R, Alqudah A, Jerotic D, Suvakov S, Obradovic D, Hoch D et al (2020) Role of a novel angiogenesis FKBPL-CD44 pathway in preeclampsia risk stratification and mesenchymal stem cell treatment. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa403
McClements L, Rayner B, Alqudah A, Grieve D, Robson T (2020) FKBPL, a novel player in cardiac ischaemia and fibrosis. J Mol Cell Cardiol 140:5. https://doi.org/10.1016/J.YJMCC.2019.11.008
Januszewski AS, Watson CJ, O’Neill V, McDonald K, Ledwidge M, Robson T, Jenkins AJ, Keech AC, McClements L (2020) FKBPL is associated with metabolic parameters and is a novel determinant of cardiovascular disease. Sci Rep. https://doi.org/10.1038/S41598-020-78676-6
Bao Y, Wenyang H, Guo Y, Yang W (2019) Phenotypic characterization of malignant progenitor cells in patients with idiopathic myelofibrosis. Hematol/Oncol Stem Cell Ther 12:146–154. https://doi.org/10.1016/J.HEMONC.2019.01.001
Garmezy B, Schaefer JK, Mercer J, Talpaz M (2021) A provider’s guide to primary myelofibrosis: pathophysiology, diagnosis, and management. Blood Rev 45:100691. https://doi.org/10.1016/j.blre.2020.100691
Giraudier S, Chagraoui H, Komura E, Barnache S, Blanchet B, LeCouedic JP, Smith DF et al (2002) Overexpression of FKBP51 in idiopathic myelofibrosis regulates the growth factor independence of megakaryocyte progenitors. Blood 100:2932–2940. https://doi.org/10.1182/blood-2002-02-0485
Komura E, Chagraoui H, Mas VMD, Blanchet B, De Sepulveda P, Larbret F, Larghero J et al (2003) Spontaneous STAT5 activation induces growth factor independence in idiopathic myelofibrosis: Possible relationship with FKBP51 overexpression. Exp Hematol 31:622–630. https://doi.org/10.1016/S0301-472X(03)00085-7
Chiasson VL, Jones KA, Kopriva SE, Mahajan A, Young KJ, Mitchell BM (2012) Endothelial cell transforming growth factor-β receptor activation causes tacrolimus-induced renal arteriolar hyalinosis. Kidney Int 82:857–866. https://doi.org/10.1038/KI.2012.104
Jiang X, Sung YK, Tian W, Qian J, Semenza GL, Nicolls MR (2014) Graft microvascular disease in solid organ transplantation. J Mol Med 92:797–810. https://doi.org/10.1007/S00109-014-1173-Y/FIGURES/2
Rodrigues-Diez R, González-Guerrero C, Ocaña-Salceda C, Rodrigues-Diez RR, Egido J, Ortiz A, Ruiz-Ortega M, Ramos AM (2016) Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci Rep 6:1–16. https://doi.org/10.1038/srep27915
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
LM and AA contributed to the study conception. RA, EQ, MW and MO did the search for the published papers. EQ and MW prepared the figures. LM and AA prepared the table. The first draft of the manuscript was written by AA and RA. LM did an extensive revision on the first draft. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alqudah, A., AbuDalo, R., Qnais, E. et al. The emerging importance of immunophilins in fibrosis development. Mol Cell Biochem 478, 1281–1291 (2023). https://doi.org/10.1007/s11010-022-04591-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04591-1