
Abstract
Apelin participates in cardiovascular functions, metabolic disease, and homeostasis disorder. However, the biological function of apelin in liver diseases, especially liver fibrosis is still under investigation. The present study aimed to investigate the expression of apelin in nonalcoholic fatty liver disease (NAFLD) and the mechanism of apelin promoting hepatic fibrosis through ERK signaling in hepatic stellate LX-2 cells. The results showed that the ALT and AST levels in serum were increased in the mice fed HFC. The histological staining revealed that hepatocellular steatosis and ballooning degeneration was severe, and fibrogenesis appeared as increased pericellular collagen deposition along with pericentral (lobular) collagen deposition in the mice fed HFC. Immunochemistry and qRT-PCR results showed that the expression of apelin and profibrotic genes was higher as compared to the control group. The in vitro experiments demonstrated that apelin-13 upregulated the transcription and translation levels of collagen type I (collagen-I) and α-smooth muscle actin (α-SMA) in LX-2 cells. The immunofluorescent staining, qRT-PCR, and Western blot results showed that the overexpression of apelin markedly increased the expression of α-SMA and cyclinD1. The LX-2 cells treated with apelin-13 displayed an increased expression of pERK1/2 in a time-dependent manner, while the pretreatment with PD98059 abolished the apelin-induced expression of α-SMA and cyclinD1. Furthermore, the in vivo and in vitro assays suggested a key role of apelin in promoting liver fibrosis, and the underlying mechanism might be ascribed to the apelin expression of profibrotic genes via ERK signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common form of chronic liver diseases in adults and children worldwide with a continual increase in the number of incidences [1]. Irrespective of the cause, liver diseases typically experience three major events, such as hepatic fibrosis, cirrhosis, and hepatocellular carcinoma. The severity of these events determined the prognosis and treatment of liver diseases. Previous studies confirmed that activated hepatic stellate cells (HSCs) are vital contributors to the fibrosis of the damaged liver [2, 3]. The activated HSCs are primarily responsible for the production of extracellular matrix (ECM) and the development of fibrosis in the liver. In the normal liver, quiescent HSCs (qHSCs) contain cytoplasmic lipid droplets primarily consisting of retinyl esters. Moreover, in response to injury or inflammatory stimulus, qHSCs are transformed to myofibroblast-like cells, which highly express the α-smooth muscle actin (α-SMA) and collagen type I (collagen-I) [4, 5]. Consequently, the activated HSCs exhibited high proliferation and migration in the liver injury sites.
Apelin is an endogenous ligand for the APJ receptor (angiotensin II receptor-like-1, AT-1). The prepro-apelin consisted of 77 amino acids that could be cleaved into several biologically active forms of peptides, such as apelin-13, -16, -17, and -36. Each isoform harbored a potent activator for APJ. Both apelin-13 and apelin-17 exerted a stronger activity than apelin-36 [6,7,8]. Furthermore, apelin played pivotal and various roles in the physiological and pathophysiological processes, including regulation of blood pressure, cardiac contractility, angiogenesis, metabolic balance, cell proliferation, apoptosis, and inflammation [9,10,11,12]. In addition, it has been shown that in the liver, apelin participated in hepatocyte apoptosis, glycogen synthesis, and fibrosis formation [13, 14]. A recent clinical investigation reported that serum apelin was associated with the histological and hemodynamic states of chronic liver disease [15]. In addition, previous studies demonstrated that hypothalamic apelin regulated the hepatic glucose metabolism in mice fed a high-fat diet [16], and high levels of serum apelin-12 were observed in human NAFLD [17]. These clinical studies suggested that apelin was positively correlated with the homeostasis model assessment (HOMA) index and body mass index (BMI) in metabolic liver disease. Also, the expression of apelin was increased sharply in the hepatic tissue of cirrhotic rats or humans [4]. Interestingly, apelin was highly expressed under hypoxic or proinflammatory conditions in HSCs, and it might promote liver fibrosis or cirrhosis progression [18, 19]. Some studies demonstrated that apelin bound to APJ led to the phosphorylation of extracellular signal-regulated kinase (ERK), protein kinase B (Akt), and p70S6 kinase signaling pathways in cell proliferation or migration activities [20,21,22]. However, whether the apelin could promote liver fibrosis by ERK signaling pathway remains elusive.
In the present study, we aimed to investigate the function and mechanism of apelin in promoting hepatic fibrosis in LX-2 cells and the mice fed high-fat chow (HFC).
Materials and methods
Animal experiments
C57BL/6 mice (SPF, males, 6–8-week-old, 22–24 g) were purchased from the Institutional Animal Care and Use Committee of Charles River (Beijing, China, License number: SCXK 2012-0001). All procedures performed in studies involving animals were carried out in accordance with the United States NIH guidelines (Guide for the Care and Use of Laboratory Animals (1985), DHEW Publication No. (NIH) 85–23: Office of Science and Health Reports, DRR/NIH, Bethesda, MD, USA) and the ethical standards of Animal Research Ethics Committee of Henan University (The experimental number: HUSOM-2017-200). The mice were divided into two groups (n = 12), the model group was fed HFC that contained 45% fat (MD12032, Medicine Led, Yangzhou, China) for 24 weeks, while the normal mice were fed a diet containing 10% fat (MD12031, Medicine Led) for the same length of time. The diet consumption and body weight were recorded weekly. At the end of 24 weeks of feeding, the mice were sacrificed, blood was collected and centrifuged, and the serum was stored at − 80 °C for biochemical analyses. The livers were harvested, snap frozen for further analysis, such as PCR and Western blotting, or stored in 10% formalin for histopathology staining.
Cell culture and treatment
LX-2 cell line was purchased from Meixuan Biological Science and Technology Ltd (Shanghai, China). The cells were seeded in 60-mm plates to confluency for 24 h in high glucose DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% FBS (fetal calf serum) in a humidified atmosphere with 5% CO2 at 37 °C. Subsequently, LX-2 cells were cultured in serum-free DMEM for 24 h, followed by treatment with apelin-13 (100 nM) (Sigma–Aldrich) for the indicated time points.
Hematoxylin–eosin (H&E) staining
The liver tissues obtained from the mice were fixed with 10% neutral-buffered formalin at room temperature for 24 h, embedded in paraffin, sectioned to a thickness of approximately 5 μm, and stained with H&E according to the standard protocol and examined by light microscopy.
Sirius red staining
The liver tissue sections were deparaffinized and hydrated in distilled water. Picrosirius red solution was applied to cover the tissue section and incubated for 60 min. Subsequently, the slides were rinsed with an acetic acid solution, followed by dehydration with absolute alcohol. Finally, the slide was mounted in synthetic resin.
Oil red-O staining
Frozen liver tissues were cut into 7 mm, then, fixed with 10% (v/v) formaldehyde for 15 min at room temperature. After washing three times with double-distilled water (ddH2O), the slides were incubated with filtered Oil Red O solution at room temperature for another 15 min, followed by further washing five times with ddH2O to remove the background staining. The liver tissue sections were observed and photographed randomly under the microscope.
Biochemical analyses
The blood of mice was collected in a common tube from an angular vein after the animals were executed, and allowed to stand for 4–6 h at room temperature, followed by centrifugation at 3000×g for 5 min. The serum was collected, and serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and cholesterol levels were determined by Reitman Frankel method using commercially available kits (Jiancheng Bioengineering Institute, Nanjing, China).
Immunohistochemistry (IHC)
The liver tissues of mice were fixed with 10% neutral-buffered formalin at room temperature for 24 h, embedded in paraffin, sectioned to a thickness of approximately 5 μm, and immunostained by a standard protocol. The immunostaining of the sections was performed using anti-apelin (GeneTex), anti-APJ (Abcam), anti-α-SMA (Abcam), and anti-cyclinD1 (Abcam) antibodies (1:100) and counterstained with hematoxylin. The staining intensities were determined by measuring the IOD (integrated optical density) with light microscopy using the computer-based Image-Pro Morphometric System in a double-blind manner.
RNA preparation and qRT-PCR (quantitative reverse transcription-PCR)
Total RNA was isolated with TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions. RNA concentrations and purity were determined by measuring the absorbance A260–A280 nm ratio. As an internal control, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene primers were used for RNA template normalization. Quantitative PCR of the related genes was performed using a Platinum SYBR Green qPCR SuperMix UDG Kit (Invitrogen). The following primers were used: apelin (mouse), 5′TCTTGGCTCTTCCCTCTTTTCA 3′ (sense) and 5 5′GTGCTGGAATCCACTGGAGAA 3′ (antisense); APJ (mouse), 5′TCGGCTAAGGCTGCGAGTC 3′ (sense) and 5′CGTCTGTGGAACGGAACAC 3′ (antisense); α-SMA (mouse), 5′GTCCCAGACATCAGGGAGTAA 3′ (sense) and 5′TCGGATACTTCAGCGTCAGGA 3′ (antisense); collagen-I (mouse), 5′ GCTCCTCTTAGGGGCCACT3′ and 5′ CCACGTCTCACCATTGGGG3′; TGF-β1(mouse), 5′ CTCCCGTGGCTTCTAGTGC3′(sense) and 5′GCCTTAGTTTGGACAGGATCTG 3′(antisense); IL-10(mouse), 5′ TAACTGCACCCACTTCCCAG3′(sense) and 5′AGGCTTGGCAACCCAAGTAA 3′ (antisense); GAPDH (mouse), 5′TGTGAACGGATTTGGCCGTA3′ (sense) and 5′ACTGTGCCGTTGAATTTGCC3′ (antisense); apelin (human) 5′, GCTCTGGCTCTCCTTGACC3′ (sense) and 5′CCATTCCTTGACCCTCTGG 3′ 3′(antisense); collagen-I (human), 5′ GAGGGCCAAGACGAAGACATC3′ (sense) and 5′ CAGATCACGTCATCGCACAAC3′ (antisense); α-SMA (human), 5′GTGTTGCCCCTGAAGAGCAT 3′ (sense) and 5′GTGTTGCCCCTGAAGAGCAT 3′ (antisense); GAPDH (human), 5′GGAGCGAGATCCCTCCAAAAT3′ (sense) and 5′GGCTGTTGTCATACTTCTCATGG3′ (antisense); The relative expression level was calculated using the following equation:relative gene expression = 2−∆∆CT.
Western blot
Crude proteins were extracted from liver tissues of mice or LX-2 cells as described previously [23, 24], resolved by SDS/PAGE and transferred on to a PVDF membrane (Millipore). Membranes were blocked with 5% (w/v) non-fat dried skimmed milk powder in TTBS (100 mM Tris/HCl, pH 7.5, 150 mM NaCl and 0.5% Tween 20) for 2 h at 37 °C and then incubated overnight at 4 °C with the following primary antibodies: 1:300 dilution rabbit anti-apelin (GeneTex), 1:1000 dilution rabbit anti-α-SMA (Abcam), 1:2500 dilution rabbit anti-cyclinD1 (Abcam), anti-β-actin and rabbit anti-IgG (Santa Cruz Biotechnology) antibodies. After incubation with the appropriate secondary antibody, the immunoreactive signal of antibody-antigens were visualized using the Chemiluminescence plus Western blot analysis kit (Tanon Biotechnology).
Immunofluorescent staining
LX-2 cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100, incubated with anti-α-SMA antibody, and further stained with Cy3-conjugated secondary antibody. Staining of 4′,6-diamidino-2-phenylindole (DAPI) was used to visualize nuclear localization. Each sections was observed under an inverted fluorescence microscope (Leica).
Plasmids transfection
Human apelin expression plasmids were purchased from Origene Technologies Inc. The transfection was performed using Lipofectamine™ reagent (Invitrogen) following the manufacturer’s instructions. At 24 h following transfection, LX-2 cells were treated with or without apelin (100 nM). Then, the cells were harvested and used for RT-PCR and Western blot.
SiRNA transfection
Small interfering RNA (siRNA)-targeting human apelin (si-apelin) and nonspecific siRNA (si-Con) were purchased from Santa Cruz Biotechnology. Transfection was performed using a Lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions. At 24 h posttransfection, LX-2 cells were harvested and used for qRT-PCR and Western blot assays.
The detection of ERK signaling
LX-2 cells were treated with 100 nM apelin-13 for the indicated time points. Total cell lysates were analyzed by Western blot using antibodies against p-ERK and ERK. LX-2 cells were pretreated with PD98059 (25 mmol/L) for 2 h, followed by a 24 h incubation with or without apelin-13 (100 nM). Subsequently, the cells were collected, and the expression of α-SMA and cyclinD1 proteins was examined by Western blot using the respective antibodies.
Statistical analyses
Data are presented as histograms of mean ± SD (standard deviation) from three or more independent experiments. Statistical analyses were performed using the Student’s t test or one-way ANOVA and post hoc test according to the number of groups compared. P < 0.05 indicated statistically significant differences.
Result
Morphological staining of the liver tissues and the biochemical analysis in NAFLD of the mice
In this study, liver fibrosis was induced in mice through continuous feeding of HFC for 24 weeks. H&E staining of the liver sections from the model mice revealed excessive steatosis and ballooning degeneration in the disorganized hepatocytes, as well as the disordered hepatic lobule structure in the surrounding portal vein (Fig. 1a). Oil red staining showed lipid accumulation and that droplets accumulated remarkably in the liver tissues of the model mice as compared to the control group (Fig. 1b). Fibrosis appeared as increased pericellular collagen deposition along with pericentral (lobular) collagen deposition in Sirius red-stained sections of model mice liver tissues (Fig. 1c). In addition, biochemical analysis showed that serum ALT, AST, and cholesterol levels were significantly increased in the mice fed HFC as compared to the controls (Fig. 1d–f). Taken together, these data indicated that the structure of the liver was damaged and the function of hepatocytes disturbed in NAFLD mice due to feeding HFC.

The morphological staining of the liver tissues and the biochemical analysis in the NAFLD of mice. a Representative images of liver histology (H&E staining, magnification: upper × 200, lower × 400), b Representative photomicrographs of liver oil red staining (magnification: upper × 200, lower × 400), c Representative photomicrographs of liver Sirus red staining (magnification: upper × 200, lower × 400), d, e. Serum ALT and AST levels. *P < 0.01 compared to the control group. F. Serum cholesterol levels. *P < 0.01 compared to the control group (n = 12 in each group)
Expression of apelin, APJ, and the related profibrotic genes in NAFLD of the mice
The mRNA levels of apelin, APJ, and the profibrosis-related genes, such as α-SMA, collagen-I, TGF-β, and IL-10, (a marked proinflammatory factor) were upregulated in the model group as compared to the control mice (Fig. 2a–f). In addition, the IHC staining revealed the expression of apelin, APJ, α-SMA, and cyclinD1 in the mice of the model group as assessed by qRT-PCR, and the arrow point to specific proteins (Fig. 3a–d). These results indicated that apelin and APJ participated in the hepatic steatosis in NAFLD of mice.

The expression of apelin, APJ, and the related profibrosis genes of the liver tissues in the NAFLD of mice. a–f Total RNA was transcribed with reverse transcriptase and amplified by PCR. GAPDH was used as an internal control. The mRNA expression of apelin, APJ, α-SMA, collagen-1, TGF-β, and IL-10 was examined by qRT-PCR from the liver tissues of the mice. *P < 0.01 compared to the control group (n = 12 in each group)

The expression of apelin, APJ, α-SMA, and cyclinD1 in the IHC staining of the liver tissues in the NAFLD of mice. a–d IHC staining of apelin, APJ, α-SMA, and cyclinD1 protein in the liver tissues of the mice (n = 12 in each group; magnification: upper × 200, lower × 400). The arrow point to specific proteins
Apelin-13 increased the α-SMA and collagen-I mRNA and protein levels in LX-2 cells
LX-2 cells belong to the hepatic stellate cell (HSC) line, and activated HSCs play a critical role in liver fibrosis. To assess whether the expression of the related marked genes, α-SMA, and collagen-I, was upregulated in apelin-stimulated LX-2 cells, RT-PCR and Western blotting methods were employed to examine the expression with respect to transcription and translation as a response to apelin signaling. When LX-2 cells were treated with 100 nM apelin-13 for various time points, the level of α-SMA and collagen-I mRNA increased in a time- and dose-dependent manner (Fig. 4a–d). In addition, the paraplastic hepatocyte was one of the primary causes of liver fibrosis, and the Western blot results showed that the expression level of cyclinD1, one of the cell cycle marker proteins, apparently enhanced the apelin-13 signaling pathway (Fig. 4e, f). These results demonstrated that apelin participated in the formation of liver fibrosis in LX-2 cells.

Apelin-13 promoted α-SMA and collagen-I mRNA and protein levels in LX-2 cells. LX-2 cells were treated with apelin-13 (100 nM) for various time points or at different doses (for 24 h). a–d Total RNA was transcribed with reverse transcriptase and amplified by PCR. GAPDH was used as an internal control. *P < 0.01 compared to the control group. e, f Western blot was performed using anti-α-SMA and anti-cyclinD1 antibodies to examine the expression of α-SMA and cyclinD1 at various time points or different doses. β-actin was used as the endogenous control
Apelin played a positive regulatory role in profibrotic genes expression
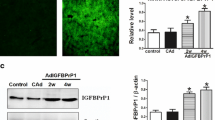
To further verify the role of apelin in regulating the expression of fibrogenesis-related genes, apelin expression plasmids were transfected in LX-2 cells to overexpress apelin. The RT-PCR and Western blot results showed that the overexpression of apelin markedly increased the expression of α-SMA and cyclinD1 at the transcription and translation levels (Fig. 5a, b). Conversely, knockdown endogenous apelin expression with si-apelin, the results showed that the expression of α-SMA and cyclinD1 markedly decreased at both mRNA and protein levels (Fig. 5c, d). Additionally, the immunofluorescent staining showed that when LX-2 cells were stimulated for 24 h by apelin-13, the α-SMA expression was distinctly upregulated (Fig. 6a). Another, the number of α-SMA (red)-positive cells was more and the expression of α-SMA was greatly increased in apelin-overexpressed LX-2 cells as compared to that of the control from the immunofluorescence results (Fig. 6b). These results suggested that apelin positively regulated the α-SMA expression in LX-2 cells.

Overexpression or knockdown of apelin regulated α-SMA mRNA and protein levels in LX-2 cells. a, b Apelin expression plasmids were transfected via Lipofectamine™ reagent following the manufacturer’s instructions. At 24 h after transfection, LX-2 cells were treated with or without apelin-13 (100 nM). c, d LX-2 cells were transfected si-apelin or si-con for 24 h, Then, the cells were harvested and used for qRT-PCR and Western blot. Total RNA was transcribed using reverse transcriptase and amplified by PCR. GAPDH was used as an internal control. *P < 0.05 compared to the control group. Western blot was performed using anti-α-SMA and anti-cyclinD1 antibodies to examine the protein expression. β-actin was used as the loading control

The effects of apelin on immunofluorescent staining of LX-2 cells. a LX-2 cells were treated with apelin-13 (100 nM) or b LX-2 cells were transfected si-apelin for 24 h, then followed by fixation in 4% paraformaldehyde, permeabilization with 0.1% Triton X-100, incubation with anti-α-SMA primary antibody, and further staining with the Cy3-conjugated secondary antibody. DAPI staining was utilized to visualize the nuclear localization. Each section was observed under an inverted fluorescence microscope
Apelin-promoted α-SMA expression through ERK signaling in LX-2 cells
The effect of apelin-13 on the expression of ERK1/2 and pERK1/2 was demonstrated by Western blot analysis. Figure 7a, c showed that the level of pERK1/2 was increased within 45 min and persisted up to 1 h after apelin-13 treatment. To further verify the role of ERK signaling in regulating the expression of profibrotic genes, LX-2 cells were pretreated with the ERK inhibitor, PD98059, for 2 h before exposure to apelin-13. Consequently, the pharmacological inhibition of ERK blocked the apelin-induced increase in α-SMA and cyclinD1 levels (Fig. 7b, d, e). These results suggested that ERK signaling pathway mediated the apelin-induced expression of profibrogenic genes in LX-2 cells (Fig. 7f).

Apelin promoted the expression of α-SMA through ERK signaling in LX-2 cells. a LX-2 cells were treated with 100 nM apelin-13 for the indicated time points. Total cell lysates were analyzed by Western blot using antibodies against p-ERK and ERK. β-actin was used as the endogenous control. b LX-2 cells were pretreated with PD98059 (25 mmol/L) for 2 h, followed by a 24 h incubation with or without apelin-13 (100 nM). Subsequently, the cells were collected, and the expression of α-SMA and cyclinD1 proteins was examined by Western blot using the respective antibodies. β-actin was used the endogenous control. c–e The densitometry analysis of the band intensities was quantized by Photoshop Image Tanon software. *P < 0.05 versus control group. f The pattern graph of apelin promotion the expression of α-SMA through ERK signaling
Discussion
NAFLD pathogenesis is associated with inflammation, steatosis, and fibrosis of chronic liver injury [25,26,27]. The present study established the genetically unaltered C57BL/6J mice model on HFC for 24 weeks. The results showed that ballooning degeneration and steatosis, as well as, lipid accumulation and fibrosis made a remarkable appearance in the liver tissues of the model mice (Fig. 1a). Furthermore, the mRNA expression of profibrotic-related genes, α-SMA, collagen-I, TGF-β, and IL-10 (Fig. 2c–f), was markedly increased in this mice model, and the results were consistent with the pathogenesis of NAFLD. Previous studies had confirmed a series different of cell signaling pathways in the pathogenesis of NAFLD [28,29,30,31]. However, the etiology of NAFLD is yet elusive. The present study demonstrated that the expression of apelin/APJ was increased in the mice fed HFC, and apelin-13 promoted hepatic fibrosis by ERK signaling in LX-2 cells.
It had been demonstrated that apelin played a vital role in the pathophysiology of many diseases, including cardiovascular disease, renal disease, type 2 diabetes, and tumors [11, 32,33,34,35]. Apelin is highly expressed in the lung, heart, mammary glands, brain, kidney, testes, and ovaries [7, 36]; however, the expression was low in normal liver [20]. A previous study reported that the apelin expression was increased sharply in the hepatic tissue of cirrhotic rats than that in the controls [4]. Furthermore, the level of circulating apelin was markedly increased in rats with cirrhosis than that in the controls. In the APJ−/− mice, the liver apoptosis and injury was significantly alleviated as compared to that in the wild-type mice [37]. The current results demonstrated that the expression of apelin and APJ was distinctly elevated in the liver tissues of the mice fed HFC than that in the control mice (Fig. 2a, b). Some studies reported that the level of serum apelin was increased in some liver diseases, such as NAFLD and cirrhosis [15,16,17]. These results suggested that apelin participated in the hepatic steatosis or fibrosis in NAFLD.
Interestingly, liver fibrosis was characterized by the excessive deposition of fibrillar collagen, especially collagen I, collagen III, and α-SMA. Therefore, the accumulation of activated HSCs was inhibited by modulating their stimulation or proliferation or promoting apoptosis, which is the leading target in preventing hepatic fibrosis [38]. The in vitro study demonstrated that apelin-13 increased the α-SMA and collagen-I mRNA and protein levels in LX-2 cells in a time- and dose-dependent manner (Fig. 4a–d). The immunofluorescent staining revealed an increased expression of α-SMA in apelin-13 stimulated LX-2 cells (Fig. 6a). The paraplastic hepatocyte is one of the main causes of liver fibrosis; the expression of cyclinD1 was increased in the apelin-13-treated LX-2 cells (Fig. 4e, f). The results demonstrated that the overexpression or knockdown of apelin markedly increased or decreased the expression of α-SMA and cyclinD1 at the transcriptional and translation levels in LX-2 cells (Fig. 5a–d), thereby rendering its role in promoting the proliferation and inducing fibrogenesis in the LX-2 cells. This phenomenon was consistent with that of the previous studies, where in apelin exerted a key role in regulating cell proliferation and apoptosis. Some studies also confirmed that the expression of apelin and APJ was low in the liver tissues of normal rats or human as compared to that in the case of cirrhosis [4]. The study demonstrated that AngII and ET-1 might exert some of their profibrotic effects in cirrhosis by activating the apelin signaling pathway [19]. However, the mechanism underlying the apelin-promoted liver profibrogenesis is yet unknown. The studies also confirmed that apelin interacted with APJ to promote the phosphorylation of p70S6K, and in turn, stimulated two signaling cascades—ERK and PI3K pathways [39, 40]. To gain further insight into the molecular mechanisms underlying the apelin-induced profibrogenesis and proliferation, as well as the α-SMA and cyclinD1 expression in LX-2 cells, we examined the intracellular signaling pathways. In the present study, we found that apelin-13 increased the expression of pERK1/2 in 45 min that persisted up to 1 h in LX-2 cells. In addition, PD98059, a pharmacological inhibitor of ERK, blocked the apelin-induced increase in α-SMA and cyclinD1 levels in LX-2 cells (Fig. 7a–e). These results suggested that ERK signaling pathway mediated the expression of apelin-induced profibrogenic and proliferation genes in LX-2 cells.
In summary, the current results showed that the expression of apelin/APJ was obviously increased in the mice fed HFC. Furthermore, apelin induced the profibrogenic and proliferation via ERK signaling pathways in LX-2 cells. Therefore, this study extended the knowledge in the field regarding the role of apelin in the development of liver fibrosis.
References
Smith BW, Adams LA (2011) Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci 48:97–113. https://doi.org/10.3109/10408363.2011.596521
Venturi C, Reding R, Quinones JA, Sokal E, Rahier J, Bueno J, Sempoux C (2016) Relevance of activated hepatic stellate cells in predicting the development of pediatric liver allograft fibrosis. Liver Transpl 22:822–829. https://doi.org/10.1002/lt.24412
Yin C, Evason KJ, Asahina K, Stainier DY (2013) Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest 123:1902–1910. https://doi.org/10.1172/JCI66369
Principe A, Melgar-Lesmes P, Fernandez-Varo G, del Arbol LR, Ros J, Morales-Ruiz M, Bernardi M, Arroyo V, Jimenez W (2008) The hepatic apelin system: a new therapeutic target for liver disease. Hepatology 48:1193–1201. https://doi.org/10.1002/hep.22467
Chen Y, Ou Y, Dong J, Yang G, Zeng Z, Liu Y, Liu B, Li W, He X, Lan T (2018) Osteopontin promotes collagen I synthesis in hepatic stellate cells by miRNA-129-5p inhibition. Exp Cell Res 362:343–348. https://doi.org/10.1016/j.yexcr.2017.11.035
De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C (2004) Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci 101:10464–10469. https://doi.org/10.1073/pnas.0403518101
Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S, Nishizawa N, Kitada C, Onda H, Nishimura O, Fujino M (2001) Molecular properties of apelin: tissue distribution and receptor binding. Biochim Biophys Acta 1538:162–171
Chaves-Almagro C, Castan-Laurell I, Dray C, Knauf C, Valet P, Masri B (2015) Apelin receptors: from signaling to antidiabetic strategy. Eur J Pharmacol 763:149–159. https://doi.org/10.1016/j.ejphar.2015.05.017
Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R, Upton PD, Crosby A, Sawiak SJ, Carpenter TA, Glen RC, Morrell NW, Maguire JJ, Davenport AP (2017) Elabela/Toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135:1160–1173. https://doi.org/10.1161/CIRCULATIONAHA.116.023218
Gerbier R, Alvear-Perez R, Margathe JF, Flahault A, Couvineau P, Gao J, De Mota N, Dabire H, Li B, Ceraudo E, Hus-Citharel A, Esteoulle L, Bisoo C, Hibert M, Berdeaux A, Iturrioz X, Bonnet D, Llorens-Cortes C (2017) Development of original metabolically stable apelin-17 analogs with diuretic and cardiovascular effects. FASEB J 31:687–700. https://doi.org/10.1096/fj.201600784R
Zhong JC, Zhang ZZ, Wang W, McKinnie SMK, Vederas JC, Oudit GY (2017) Targeting the apelin pathway as a novel therapeutic approach for cardiovascular diseases. Biochim Biophys Acta Mol Basis Dis 1863:1942–1950. https://doi.org/10.1016/j.bbadis.2016.11.007
Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C, Zhu CH, Miao JY (2016) Apelin-13 protects against apoptosis by activating AMP-activated protein kinase pathway in ischemia stroke. Peptides 75:96–100. https://doi.org/10.1016/j.peptides.2015.11.002
Lv X, Kong J, Chen WD, Wang YD (2017) The role of the apelin/APJ system in the regulation of liver disease. Front Pharmacol 8:221. https://doi.org/10.3389/fphar.2017.00221
Chu J, Zhang H, Huang X, Lin Y, Shen T, Chen B, Man Y, Wang S, Li J (2013) Apelin ameliorates TNF-alpha-induced reduction of glycogen synthesis in the hepatocytes through G protein-coupled receptor APJ. PLoS ONE 8:e57231. https://doi.org/10.1371/journal.pone.0057231
Lim YL, Choi E, Jang YO, Cho YZ, Kang YS, Baik SK, Kwon SO, Kim MY (2016) Clinical implications of the serum apelin level on portal hypertension and prognosis of liver cirrhosis. Gut Liver. https://doi.org/10.5009/gnl14345
Drougard A, Duparc T, Brenachot X, Carneiro L, Gouaze A, Fournel A, Geurts L, Cadoudal T, Prats AC, Penicaud L, Vieau D, Lesage J, Leloup C, Benani A, Cani PD, Valet P, Knauf C (2014) Hypothalamic apelin/reactive oxygen species signaling controls hepatic glucose metabolism in the onset of diabetes. Antioxid Redox Signal 20:557–573. https://doi.org/10.1089/ars.2013.5182
Ercin CN, Dogru T, Tapan S, Kara M, Haymana C, Karadurmus N, Karslioglu Y, Acikel C (2010) Plasma apelin levels in subjects with nonalcoholic fatty liver disease. Metabolism 59:977–981. https://doi.org/10.1016/j.metabol.2009.10.019
Yokomori H, Oda M, Yoshimura K, Machida S, Kaneko F, Hibi T (2011) Overexpression of apelin receptor (APJ/AGTRL1) on hepatic stellate cells and sinusoidal angiogenesis in human cirrhotic liver. J Gastroenterol 46:222–231. https://doi.org/10.1007/s00535-010-0296-3
Melgar-Lesmes P, Casals G, Pauta M, Ros J, Reichenbach V, Bataller R, Morales-Ruiz M, Jimenez W (2010) Apelin mediates the induction of profibrogenic genes in human hepatic stellate cells. Endocrinology 151:5306–5314. https://doi.org/10.1210/en.2010-0754
D’Aniello C, Lonardo E, Iaconis S, Guardiola O, Liguoro AM, Liguori GL, Autiero M, Carmeliet P, Minchiotti G (2009) G protein-coupled receptor APJ and its ligand apelin act downstream of Cripto to specify embryonic stem cells toward the cardiac lineage through extracellular signal-regulated kinase/p70S6 kinase signaling pathway. Circ Res 105:231–238. https://doi.org/10.1161/CIRCRESAHA.109.201186
Shin K, Chapman NA, Sarker M, Kenward C, Huang SK, Weatherbee-Martin N, Pandey A, Dupre DJ, Rainey JK (2017) Bioactivity of the putative apelin proprotein expands the repertoire of apelin receptor ligands. Biochim Biophys Acta Gen Subj 1861:1901–1912. https://doi.org/10.1016/j.bbagen.2017.05.017
Zhang ZZ, Wang W, Jin HY, Chen X, Cheng YW, Xu YL, Song B, Penninger JM, Oudit GY, Zhong JC (2017) Apelin is a negative regulator of angiotensin II-mediated adverse myocardial remodeling and dysfunction. Hypertension 70:1165–1175. https://doi.org/10.1161/HYPERTENSIONAHA.117.10156
Zheng B, Han M, Shu YN, Li YJ, Miao SB, Zhang XH, Shi HJ, Zhang T, Wen JK (2011) HDAC2 phosphorylation-dependent Klf5 deacetylation and RARalpha acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. https://doi.org/10.1038/cr.2011.34
Lv XR, Zheng B, Li SY, Han AL, Wang C, Shi JH, Zhang XH, Liu Y, Li YH, Wen JK (2013) Synthetic retinoid Am 80 up-regulates apelin expression by promoting interaction of RARalpha with KLF5 and Sp1 in vascular smooth muscle cells. Biochem J 456:35–46. https://doi.org/10.1042/BJ20130418
Baciu C, Pasini E, Angeli M, Schwenger K, Afrin J, Humar A, Fischer S, Patel K, Allard J, Bhat M (2017) Systematic integrative analysis of gene expression identifies HNF4A as the central gene in pathogenesis of non-alcoholic steatohepatitis. PLoS ONE 12:e0189223. https://doi.org/10.1371/journal.pone.0189223
Ichimura M, Masuzumi M, Kawase M, Sakaki M, Tamaru S, Nagata Y, Tanaka K, Suruga K, Tsuneyama K, Matsuda S, Omagari K (2017) A diet-induced Sprague-Dawley rat model of nonalcoholic steatohepatitis-related cirrhosis. J Nutr Biochem 40:62–69. https://doi.org/10.1016/j.jnutbio.2016.10.007
Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, Fang C, Lu YX, Yang X, Gao MM, Zhang Y, Tian S, Zhu XY, Gong J, Ma XL, Li F, Wang Z, Huang Z, She ZG, Li H (2018) The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med 24:84–94. https://doi.org/10.1038/nm.4453
Woolbright BL, Jaeschke H (2017) Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol 66:836–848. https://doi.org/10.1016/j.jhep.2016.11.017
Chung RT, Stravitz RT, Fontana RJ, Schiodt FV, Mehal WZ, Reddy KR, Lee WM (2012) Pathogenesis of liver injury in acute liver failure. Gastroenterology 143:e1–e7. https://doi.org/10.1053/j.gastro.2012.07.011
Ramaiah SK, Jaeschke H (2007) Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol 35:757–766. https://doi.org/10.1080/01926230701584163
Hu H, He L, Li L, Chen L (2016) Apelin/APJ system as a therapeutic target in diabetes and its complications. Mol Genet Metab 119:20–27. https://doi.org/10.1016/j.ymgme.2016.07.012
Han X, Wang LY, Diao ZL, Liu WH (2016) Apelin: a novel inhibitor of vascular calcification in chronic kidney disease. Atherosclerosis 244:1–8. https://doi.org/10.1016/j.atherosclerosis.2015.10.102
Bertrand C, Valet P, Castan-Laurell I (2015) Apelin and energy metabolism. Front Physiol 6:115. https://doi.org/10.3389/fphys.2015.00115
Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, Merchant AS, Mehta GU, Chichura A, Shalem O, Tran E, Eil R, Sukumar M, Guijarro EP, Day CP, Robbins P, Feldman S, Merlino G, Zhang F, Restifo NP (2017) Identification of essential genes for cancer immunotherapy. Nature 548:537–542. https://doi.org/10.1038/nature23477
Muto J, Shirabe K, Yoshizumi T, Ikegami T, Aishima S, Ishigami K, Yonemitsu Y, Ikeda T, Soejima Y, Maehara Y (2014) The apelin-APJ system induces tumor arteriogenesis in hepatocellular carcinoma. Anticancer Res 34:5313–5320
Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, Kitada C, Honda S, Kurokawa T, Onda H, Nishimura O, Fujino M (2000) Molecular and functional characteristics of APJ tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem 275:21061–21067. https://doi.org/10.1074/jbc.M908417199
Yasuzaki H, Yoshida S, Hashimoto T, Shibata W, Inamori M, Toya Y, Tamura K, Maeda S, Umemura S (2013) Involvement of the apelin receptor APJ in Fas-induced liver injury. Liver Int 33:118–126. https://doi.org/10.1111/liv.12006
Schuppan D, Kim YO (2013) Evolving therapies for liver fibrosis. J Clin Invest 123:1887–1901. https://doi.org/10.1172/JCI66028
Masri B, Morin N, Cornu M, Knibiehler B, Audigier Y (2004) Apelin (65–77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J 18:1909–1911. https://doi.org/10.1096/fj.04-1930fje
Rostamzadeh F, Najafipour H, Yeganeh-Hajahmadi M, Esmaeili-Mahani S, Joukar S, Iranpour M (2017) Heterodimerization of apelin and opioid receptors and cardiac inotropic and lusitropic effects of apelin in 2K1C hypertension: role of pERK1/2 and PKC. Life Sci 191:24–33. https://doi.org/10.1016/j.lfs.2017.09.044
Acknowledgements
This study was supported by National Natural Science Foundation of China (Grant Nos. U1404310, 81600940 and 81600974) and Foundation of Science & Technology Department of Henan Province, China (No. 182102310371).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Y., Song, J., Bian, H. et al. Apelin promotes hepatic fibrosis through ERK signaling in LX-2 cells. Mol Cell Biochem 460, 205–215 (2019). https://doi.org/10.1007/s11010-019-03581-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-019-03581-0