
Abstract
West Nile virus (WNV) is a major pathogenic flavivirus which causes human neuro-invasive disease, worldwide. Still successful vaccine and therapeutic treatment against WNV infection is not available, which demands the development of more potential WNV vaccines. The present study used immunoinformatics methods viz. Matrix and Artificial Neural Network (ANN) based algorithm to identify the promiscuous and conserve T cell epitopes from entire WNV proteome followed by structure based analysis of identified epitopes. The epitopes were also taken for TAP binding analysis and epitope conservancy analysis. Among 89 identified epitopes, eight epitopes showed high potential and conserve nature but two epitopes viz. capsid 40FVLALLAFF48 and NS2B 9LMFAIVGGL17 were found most promiscuous and having high population coverage in comparison of other identified epitopes and known antigenic positive control epitopes. Further, Autodock 4.2 and NAMD–VMD molecular dynamics simulation were used for docking and molecular dynamics simulation respectively, to validate epitope and allele complex stability. The 3D structure models were generated for epitopes and corresponding HLA allele by Pepstr and Modeller 9.10, respectively. Epitope FVLALLAFF-B*3501 allele and epitope LMFAIVGGL-B*5101 HLA allele complexes have shown best energy minimization and stable complexes during simulation. The study also showed the optimum binding epitopes FVLALLAFF and LMFAIVGGL with cTAP1 (PDB ID: 1JJ7) cavity, as revealed by Autodock 4.2, concluding favored passage through the ER membrane from cytosol to the ER lumen during cytosolic processing. The docking experiment of epitopes FVLALLAFF, LMFAIVGGL with cTAP1 very well show 1 H-bond state with a binding energy of −1.62 and −0.23 kcal/mol, respectively. These results show a smooth pass through of the epitope across the channel of cTAP1 via being weakly bonded and released into the ER lumen through the cavity of cTAP1. Overall, identified peptides have potential application in the development of short peptide based vaccines and diagnostic agents for West Nile virus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
West Nile virus (WNV) is a mosquito born virus classified under the flaviviridae family comprising 72 other viruses such as yellow fever virus, Japanese encephalitis virus, dengue virus, virus and tick-born encephalitis virus. WNV is transmitted to human being by Culex mosquito species viz. Cx. pipiens, Cx. perexiguus, Cx. modestus, Cx. Tarsalis and Cx. Quinquefasciatus (Petersen et al. 2013). WNV has single stranded RNA genome of 11,000 nucleotides translating a single polypeptide of 3433 amino acids (Brinton 2014). The virus has three structural (Capsid, precursor membrane protein and envelope protein) and seven nonstructural (NS1, NS2a, NS2b, NS3, NS4a, NS4b and NS5) proteins. In human, WNV leads to cause West Nile (WN) fever and neuro-invasive diseases. As reported, 16,196 clinical cases with WNV Neuro-invasive disease have been detected from 1999 to 2012 with approximately 1549 deaths in the United States (Amanna and Slifka 2014). WN fever is characterized by several clinical symptoms such as headache, malaise, fever, myalgia, chills, vomiting, rashes, fatigue, eye pain and sometimes lymphadenopathy. In severe WNV infection cases (<1%) of WN Neuro-invasive disease is characterized by meningitis, encephalitis, acute flaccid paralysis, arthralgia, ataxia, visual disturbances, tremors, bulbar dysfunction etc. (Petersen et al. 2013). However, no human vaccine and therapeutic treatment are yet available for WNV infection (CDC 2015). Presently ribavirin, pyridazine nucleoside, corticosteroids and interferon α-2b are being used to subdue the symptoms of WN viral infection (Beasley 2011). Overall, there is an urgent need for effective and specific targeted vaccine and therapeutic means to answer the hazardous infection being proliferated by WNV. The option of epitope vaccine provides rather more effective, more specific immune response and also does not have side effects in contrast of whole virus based conventional vaccines (De Groot et al. 2002; Sharma and Kumar 2010). The conventional vaccine approaches are not viable for pathogens which are antigenically diverse and not cultivable in the laboratory (Singh and Mishra 2016). In addition to these potentials, the major benefit of epitope based vaccine is the ability to give high doses of immunogen at economical cost (Tang et al. 2012).
In acquired immune system, antigens can only be recognized by T cells when they are bound with major histocompatibility complex (MHC) (Zhang et al. 2005). MHC molecules are highly polymorphic and also known as human leukocyte antigens (HLA) in humans. HLA molecules have the ability to present a range of peptide epitopes on the surface of cells for recognition by T cells (Sharma et al. 2014). The most promiscuous T cell epitopes can bind with number of HLA supertype alleles to cover different populations which lower the chance of antigen escape related to antigenic drift or shift. The HLA supertype concept has a profound role in the perceptive of T cell peptide epitope assortment and disintegration during T cell immune responses (Kangueane et al. 2005). The immune system has two antigen processing and presentation pathways viz. cytosolic pathway and endocytic pathway. Endogenous and exogenous antigens are processed and presented on the membrane by HLA class I and II molecules under the cytosolic and endocytic pathway, respectively. The transporter associated with antigen processing (TAP) protein is involved in transport of cytosolic peptides into rough endoplasmic reticulum (RER) to further bind with HLA class I molecules during cytosolic pathway. TAP protein is present in the membrane of RER which act as a channel between cytosol and lumen of RER. Therefore peptides binding to HLA class I and TAP protein is a crucial factor to initiate an immune response (Procko and Gaudet 2009; Gaudet and Wiley 2001).
Here we present sequence and structure based study of identified promiscuous HLA and cTAP binding T cell epitopes. 3D structure models were generated for epitopes and corresponding HLA allele by Pepstr and Modeller 9.10, respectively for their structure based study. The structure based study involve the docking of the identified consensus peptide nanomer epitope with cTAP1 and favored HLA alleles to confirm the antigen processing and presentation process. The docking study of epitope with cTAP1 comes as a cross check that identified conserved epitope is indeed well bound and channeled by the cTAP1 cavity from cytoplasm to the ER lumen for HLA class I antigen processing and presentation. Further, best promiscuous epitopes analyzed for their binding stability to respective HLA allele by NAMD–VMD molecular dynamics simulation.
Methodology
Lineage 1a WN strain viruses are widely distributed in Africa, Europe, the Middle East, Asia, and the America regions (Petersen and Roehrig 2001); hence it can be used for vaccine development which can also provide cross protection against others WNV lineages. The protein sequences of WNV strain (Accession No. AGI16461) were retrieved from sequence database NCBI. For validation of the predicted epitopes as potential antigens; the Hepatitis core protein (Accession No. CAA59535) peptide epitope 141STLPETTVV149 and H1N1 Nucleoprotein (Accession No. P03466) peptide epitope 265ILRGSVAHK273 were taken as positive control (Singh et al. 2015a; Ansari et al. 2009).
Identification of T Cell Peptide Epitopes and Their Conservancy Study
All non-structural and structural proteins of WNV were examined for identification of HLA class I binding T cell peptide epitopes using immunoinformatics tools viz. Propred I. Propred I (http://crdd.osdd.net/raghava/propred1/) tool was employed to screen the all possible peptides against 47 HLA class I alleles at 4% threshold (Singh and Raghava 2003). Later Identified epitopes were analyzed for their TAP binding property by MHC Pred 2.0 (http://www.ddg-pharmfac.net/mhcpred/MHCPred/help.htm) (Guan et al. 2006) and worldwide conservancy study among different lineages/geographical regions strains by the IEDB conservancy tool (http://tools.iedb.org/conservancy). To perform the conservancy analysis, all protein sequences of all genotypes belong to different geographical regions (India, USA, Italy and Russia) was obtained randomly from NCBI database. IEDB MHC class I binding prediction tool (http://tools.iedb.org/mhci/) with recommended Artificial Neural Network (ANN) algorithm (Nielsen et al. 2003) was also employed for supertype binding analysis of highly conserved peptides for five (A2, A3, B7, A24, and B15) class I HLA supertypes. The identified nanomeric peptides with less than 50 IEDB percentile value and 88–100% conservancy with a maximum single mutation were selected for further study. This immunoinformatics approach helped in finding of promiscuous epitopes among identified T cell epitopes. Promiscuous epitopes are those epitopes which bind with all HLA allele members of HLA supertypes (Burrows et al. 2003; Fig. 1).

Flowchart of immunoinformatics top down approach employed in sequence and structure based binding prediction study of HLA class I and TAP binding peptides for WNV vaccine development (Sharma et al. 2017)
Here the study includes 20 HLA alleles of five HLA class I supertypes viz. A2 (A*0201, A*0202, A*0203, A*0205, A*0206), A3 (A*0301, A*1101, A*3101, A*3301, A*6801), A24 (A*2301, A*2402, A*2403, A*2405, A*2407), B7 (B*5101, B*5102, B*5301, B*0702, B*3501) and B15 (A*0101, B*1501, B*1502) to cover maximum population (Reche and Reinherz 2005). Further, this analysis also included the two known positive controls antigenic peptide epitopes as similar as we have taken in immunoinformatics study for Japanese encephalitis (Sharma et al. 2017). The positive controls were analyzed for their IEDB percentile value against five (A2, A3, A24, B7 and B15) class I HLA supertype alleles to compare with identified epitopes. Hepatitis core protein (Accession No. CAA59535) peptide epitope 141STLPETTVV149 and H1N1 Nucleoprotein (Accession No. P03466) peptide epitope 265ILRGSVAHK273 have taken as positive control (Singh et al. 2015a; Ansari et al. 2009).
Structural Modeling and Validation of Selected HLA Alleles and Identified Epitopes
Among all screened nanomer peptide epitopes, one epitopes of capsid protein (FVLALLAFF) and one epitope of non structural protein NS2B (LMFAIVGGL) of WNV were identified as potential epitopes on the basis of propred I screening, IEDB percentile score, high conservancy and population allele coverage. Epitopes were modeled by Pepstr (Kaur et al. 2007) and their structure also validated by Amber 6.0. All experimented HLA alleles sequences were obtained from IMGT database (Robinson et al. 2015) and modeled using corresponding templates by MODELLER9.10 (Sali 2014). HLA alleles models were further validated by using Errat (Colovos and Yeates 1993), ProSA (Wiederstein and Sippl 2007), ProQ (Wallner and Elofsson 2003) and RAMPAGE (Lovell et al. 2002).
Molecular Docking Study of Identified Epitopes with their Favored Alleles and Human cTAP1
After structural modeling of identified peptide epitopes and favored HLA alleles, molecular binding simulation was performed by Autodock4.2 and Hex8.0. (Morris et al. 2009; Ritchie et al. 2008). For docking, water molecules were removed from the receptor, added polar hydrogen to it and also necessary charges (Gasteiger and Kollman charges etc.) were added to generate final docking receptor .pdbqt file. The ligands (epitopes) were kept free for bond rotation except peptide bonds. Discovery studio visualizer tool was used for analysis of the docked peptide and allele complexes. These binding analyses were further validated by Hex 8.0 interactive molecular graphics program. The Fast Fourier transformation based Hex docking (Hex 8.0) was framed with grid dimension of 0.6 with a range of 180 and step size 7.5. After binding analysis with favored HLA alleles, best HLA class I binding epitopes were identified for docking study with human cTAP1(PDB-1JJ7) to confirm their antigenic processing during cytosolic process.
Molecular Dynamic Simulation of Epitope–HLA Allele Complexes
Nanoscale molecular dynamics (NAMD) with visual molecular dynamics (VMD) was used for molecular dynamics (MD) simulation study (James et al. 2005; Humphrey et al. 1996). In order to run MD simulations for epitope and allele complex, we generated a Protein complex structure file (PSF) by accessing .pdb files through the PSF builder tool of VMD. The file .psf was generated by using various force field parameters such as bond strengths, equilibrium lengths and various bonding interactions. Finally trajectosry .dcd file generated by NAMD. Root mean square deviation (RMSD) value of the complex was calculated by using rmsd.tcl source file from the Tk console of VMD. Finally RMSD was saved as rmsd.dat file and Microsoft Excel was used to plot the values in the file rmsd.dat. RMSD graph was generated for an equilibrated MD simulation system of epitope and allele complexes.
Result
The present study is top down Immunoinformatics based approach to identify the potential vaccine candidates in the form of T cell epitope which is divided in two parts (i) sequence based study to screen the potential T cell epitopes from whole WNV proteome (ii) structure based study for validation of screened most potential T cell epitopes to their potentiality as antigen or vaccine candidates.
Identification of HLA Class I Allele, TAP Binding Epitopes and Conservancy Study
Among all 89 identified HLA class I epitopes, only eight epitopes showed high conservancy and high potential in sequence based analysis, also from among them one structural capsid protein epitope (FVLALLAFF) and one epitope of a NS2B protein (LMFAIVGGL) were predicted most potential epitopes in terms of Propred screening, TAP binding IC50 score, conserve nature and HLA allele coverage (Table 1). The epitope LMFAIVGGL was found 100% conserve nature among all different geographical strains and also showed TAP binding property with IC50 value of 25.0 nM. But epitope FVLALLAFF showed 88% conserve nature with a single mutation (Alanine replaced by Valine amino acid) FVLALLA(V)FF and also showed TAP binding property with IC50 value of 6.71 nM. The study showed that among all identified epitopes, including positive control (Hepatitis core protein (Accession No. CAA59535) peptide epitope 141STLPETTVV149 and H1N1 Nucleoprotein (Accession No. P03466) peptide epitope 265ILRGSVAHK273) epitopes, only two epitopes viz. FVLALLAFF and LMFAIVGGL are binding to all allele members of A2, B7, A3, A24, and B15 HLA class I supertypes with less than 50 percentile rank value (Tables 1 and 2). This analysis showed that these two epitopes were also found more superior in term of high population coverage than positive controls as shown in Table 2. Therefore FVLALLAFF and LMFAIVGGL epitopes were identified as super antigenic or promiscuous. These identified super antigenic epitopes and favored HLA alleles, were modeled to analyze their binding simulations.
Structural Modeling of Identified Epitopes and HLA Alleles
Structural model of FVLALLAFF and LMFAIVGGL epitopes were generated by Pepstr, which were further refined with energy minimization and MD simulation using Amber 6.0. The IMGT/HLA Database and Protein Data Bank (PDB) Database allowed us to retrieve information upon a specific HLA allele sequences and their pdb structures. HLA class I, A*0101, A*0201, A*0301, B*0702, B*3501, B*5101 and B*5301 alleles and their PDB ID are 4NQV, 1A07, 3RL1, 3VCL, 1XH3, 1E27 and 1A10, were taken for docking experiments respectively. The B5102 allele structure was not retrieved from PDB database, will be modeled with the help of MODELLER9.10 by using template 1BII PDB. Structural model of allele B5102 was validated by using several tools viz. ProSA, Errat, Pro Q and RAMPAGE. Models quality was acceptable on the basis of OQF, LG score, Max Sub and Z score values (Fig. 2; Table 3). Residues in favored region were 91.6% on Ramachandran plot for B5102 alleles as revealed by RAMPAGE.

ProSA analysis: Z score plot of B5102 allele representing −9.15 Z score
Binding Simulation Study of Identified Peptide Epitopes and HLA Alleles
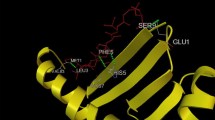
Docking study of epitopes FVLALLAFF and LMFAIVGGL with major HLA alleles of HLA class I supertypes were done to reveal their binding pattern. Binding of FVLALLAFF and LMFAIVGGL epitopes with B3501 and B5101HLA allele showed the lowest binding energy of −4.80 and −4.57 kcal/mol, respectively. Stable complex of FVLALLAFF-B3501 formed two H-bond viz. ARG357:HN1 and LEU363 (Fig. 3). Similarly, stable complex of LMFAIVGGL-B*5101 allele formed two H bond viz. with ASP30 and TRY302 (Fig. 4). Validation using Hex 8.0 also showed that FVLALLAFF-B3501 and LMFAIVGGL-B*5101 interactions formed stable complexes with one H-bond. Binding energy of epitopes with favored HLA alleles by Autodock 4.2 and Hex 8.0, are tabulated in Tables 4 and 5.

Docked epitope FVLALLAFF-B3501 allele complex by Autodock 4.2 showing detailed position of amino acids along with formation of 2 H-bonds (yellow dotes string) with LEU363 and ARG357. (Color figure online)

Docked epitope LMFAIVGGL-B5101 allele complex by Autodock 4.2 showing detailed position of amino acids along with formation of 2 H-bonds (green dotes string) with TYR302 and ASP30. (Color figure online)
Furthermore, we also present the docking study of identified highly conserved peptide epitopes FVLALLAFF and LMFAIVGGL with human cTAP1 (PDB-1JJ7) for their cytosolic processing. This docking study shows that the epitope peptides gets hold in the cavity by one hydrogen bond each, which may conclude a smooth passage for peptide epitope transport from cytoplasm to ER lumen.
Docking study of these identified epitopes with cTAP1 (PDB-1JJ7) show optimum binding with acceptable binding energy. Epitope FVLALLAFF is forming one hydrogen bond in the cavity of cTAP1 viz. ARG659 with overall binding energy −1.62 kcal/mol (Fig. 5); epitope LMFAIVGGL is forming one hydrogen bond in the cTAP1 viz. GLN552 with overall binding energy −0.23 kcal/mol (Fig. 6).

a The docking study of identified highly conserved epitope FVLALLAFF (grey solid spheres) with cTAP1 (white solid spheres) channel cavity facilitating the smooth passage of the epitope from cytoplasm to ER lumen. b This docking study shows that the FVLALLAFF epitope peptide gets hold by one hydrogen bond at cavity of cTAP1 viz. ARG659 with binding energy −1.62 kcal/mol

a The docking study of identified highly conserved epitope LMFAIVGGL (grey solid spheres) with cTAP1 (white solid spheres) channel cavity facilitating the smooth passage of the epitope from cytoplasm to ER lumen. b This docking study shows that the LMFAIVGGL epitope peptide gets hold by one hydrogen bond at cavity of cTAP1 viz. GLN552 with binding energy −0.23 kcal/mol
NAMD Simulation of Epitope–HLA Allele Complexes
The complexes of peptide and allele produced by Autodock4.2 were employed for their binding stability by NAMD-VMD simulation. Peptide FVLALLAFF-B3501 allele complex showed the highest 11 Å RMSD value (Fig. 7) likewise LMFAIVGGL-B5101 allele complex showed the highest 11.4 Å RMSD value (Fig. 8). The RMSD values of both the complexes are acceptable to conclude stable binding.

Graph displaying RMSD in relation to time (picoseconds) for NAMD-VMD simulation of WNV peptide FVLALLAFF and B3501 allele complex, with highest RMSD value of 11 Å at 7000 picoseconds

Graph displaying RMSD in relation to time (picoseconds) for NAMD-VMD simulation of WNV peptide LMFAIVGGL and B5101 allele complex, with highest RMSD value of 11.4 Å at 10,600 picoseconds
Discussion
In the presented study, Propred I and IEDB were employed for mapping of best T cell epitopes from proteome of WNV with consideration of different geographical conservancy study FVLALLAFF and LMFAIVGGL epitopes were identified as highly conserved, high propred HLA class I allele binding frequency and promiscuous with maximum population coverage among all predicted T cell epitopes.
Among all identified epitopes, including two positive control peptides, only two epitopes viz. FVLALLAFF and LMFAIVGGL of WNV were found as best epitope in terms of the HLA allele population coverage with conserve nature and were also good cTAP1 binders. TAP binding property is necessary to transport the cytosolic processed epitopes into the lumen of the RER through TAP protein to bind with MHC class I alleles. Study showed that FVLALLAFF and LMFAIVGGL epitopes binding to all allele members of A2 (A*0201, A*0202, A*0203, A*0205, A*0206), A3(A*0301, A*1101, A*3101, A*3301, A*6801), A24 (A*2301, A*2402, A*2403, A*2405, A*2407), B7 (B*5101, B*5102, B*5301, B*0702, B*3501) and B15 (A*0101, B*1501, B*1502) HLA class I supertype with less than 50 IEDB percentile value (Table 2). Therefore epitopes FVLALLAFF and LMFAIVGGL were identified as superantigenic or promiscuous peptide epitopes for WN. Altogether the above immunoinformatics analysis, hence shows that the identified peptides are most promising vaccine candidates for WNV, this was further validated by binding simulation analysis with favored HLA alleles of class I.
The binding of epitopes and HLA alleles revealed by Autodock 4.2 were further confirmed by Hex 8.0. The Hex energy of the FVLALLAFF-B3501 complex obtained by Hex 8.0 was acceptable. Autodock 4.2 results for docking of FVLALLAFF-B3501 revealed binding energy of −4.80 kcal/mol and formed two H-bonds viz. ARG357 and LEU363amino acid residues. The NAMD-VMD study further confirms that the FVLALLAFF-B3501 complex indeed attains stability with acceptable RMSD with a time frame of 7,000 picoseconds. Likewise Hex energy and binding energy of LMFAIVGGL-B5101 allele was also observed to be in the acceptable range of Hex 8.0 and Autodock 4.2 tools. Autodock result of this complex revealed the binding energy to be −4.57 kcal/mol forming two H-bonds viz. ASP30 and TRY302. The NAMD-VMD study further confirms that the LMFAIVGGL-B5101 complex indeed attains stability with acceptable RMSD with a time window of 10,600 picoseconds. In addition to the above studies of the complex of epitope and alleles, we also performed the study of docking for epitopes FVLALLAFF, LMFAIVGGL with the cTAP1 which show appropriate binding in cTAP cavity with binding energy −1.62 and −0.23 kcal/mol, respectively. This study revealed that the identified epitope is transported from cytoplasm to ER lumen through the cTAP1 cavity as shown in Figs. 5 and 6. The epitope get a hold in the cTAP1 cavity by one hydrogen bond. With the optimum binding energy in the cTAP cavity, we may conclude a smooth passage for peptide epitope transport from cytoplasm to ER lumen.
The present study has identified epitopes FVLALLAFF and LMFAIVGGL as the most promising candidate for WNV vaccine having super antigenic property. Epitope LMFAIVGGL was also found as a potential HLA class I supertype binder in our similar study for Japanese encephalitis (Sharma et al. 2017) which gives a prospect of a common epitope of multiple sources to develop a vaccine of multiple pathogens. Epitope LMFAIVGGL was found common promiscuous and conserve in both infectious WNV and JEV. These identified potential novel peptide nanomer epitopes are more effective candidates in contrast to the vaccine candidates as whole viral proteins; furthermore, it has been reported that few number of peptide epitopes can represent complete antigenicity of a protein (De Groot et al. 2002). In agreement with this immunoinformatics study, peptide epitope based vaccines have shown potential result against several highly infectious diseases such as JEV, HIV, H1N1, H7N9 and Tuberculosis (Sharma et al. 2017; Sharma and Kumar 2010, Jardine et al. 2013, De Groot et al. 2013; Feng et al. 2013). Immunoinformatics appears to be one of the fields that accelerate the immunological research progression to the development of effective vaccines (Khalili 2014). These identified epitopes could be tested as vaccine candidates and diagnostic reagents for WNV as further prospects.
Conclusion
We document the identification of HLA class I binding WN viral nanomer peptides epitopes revealed from Propred I and IEDB, respectively along with worldwide conservancy. We further found that the peptides 40FVLALLAFF48 and 9LMFAIVGGL17 are highly conserved and super antigenic epitopes. Identified capsid epitope 40FVLALLAFF48 and NS2B epitope 9LMFAIVGGL17 showed stability in complex form with B3501 and B5101 HLA alleles respectively; additionally smooth passage of these peptide epitopes through cTAP1 shows actual cytosolic antigen processing for membrane surface presentation by HLA class I. These epitopes have shown the high conservancy with sequence and structure based analysis with higher capability in comparison of positive controls as vaccine candidates. Finally manifold sequence based screening and their validation by structure based study result potential immunogenic T cell HLA class I epitopes as promising WNV vaccine candidates; no doubt candidacy of epitopes will be further tested by in vivo or in vitro methods.
Change history
27 October 2017
The original version of this article unfortunately contained an error. One of the author name Sukrit Srivastav and his affiliation details was inadvertently omitted from the author group. The author’s name and affiliation are now included in this correction.
References
Amanna IJ, Slifka MK (2014) Current trends in West Nile virus vaccine development. Expert Rev Vaccines 13(5):589–608
Ansari HR, Flower DR, Raghava GPS (2009) AntigenDB: an immunoinformatics database of pathogen antigens. Nucleic Acids Res 38:D847–D853
Beasley DW (2011) Vaccines and immunotherapeutics for the prevention and treatment of infections with West Nile virus. Immunotherapy 3(2):269–285
Brinton MA (2014) Replication cycle and molecular biology of the West Nile virus. Viruses 6(1):13–53
Burrows SR, Elkington RA, Miles JJ, Green KJ, Walker S, Haryana SM, Moss DJ, Dunckley H, Burrows JM, Khanna R (2003) Promiscuous CTL Recognition of Viral Epitopes on Multiple Human Leukocyte Antigens: Biological Validation of the Proposed HLA A24 Supertype. The Journal of Immunology 171:1407–1412
CDC (2015) West Nile virus. http://www.cdc.gov/westnile/symptoms. Accessed 31 May 2017
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of non-bonded atomic interactions. Protein Sci 2:1511–1519
De Groot AS, Sbai H, Aubin CS, McMurry J, Martin W (2002) Immuno-informatics: mining genomes for vaccine components. Immunol Cell Biol 80:255–269
De Groot AS, Ardito M, Terry F, Levitz L, Ross T, Moise L (2013) Low immunogenicity predicted for emerging avian-origin H7N9. Hum Vaccin Immunother 9(5):950–956
Feng G, Jiang Q, Xia M, Lu Y, Qiu W, Zhao D, Lu L, Peng G, Wang Y (2013) Enhanced Immune Response and Protective Effects of Nano-chitosan-based DNA Vaccine Encoding T Cell Epitopes of Esat-6 and FL against Mycobacterium Tuberculosis Infection. PLoS ONE 8(4):e61135
Gaudet R, Wiley DC (2001) Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. The EMBO Journal 20:4964–4972
Guan P, Hattotuwagama CK, Doytchinova IA, Flower DR (2006) MHCPred 2.0: an updated quantitative T-cell epitope prediction server. Appl Bioinformatics 5:55–61
Humphrey W, Dalke A, Schulten K (1996) VMD—visual molecular dynamics. J Mol Graphics 14:33–38
James CP, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, MacPherson S, Jones M, Nieusma T, Mathison J, Baker D, Ward AB, Burton DR, Stamatatos L, Nemazee D, Wilson Ia, Schief WR (2013) Rational HIV immunogen design to target specific germline B cell receptors. Science 340:711–716
Kangueane P, Sakharkar MK, Rajaseger G, Bolisetty S, Sivasekari B, Zhao B, Ravichandran M, Shapshak P, Subbiah S (2005) A framework to sub-type HLA supertypes. Front Biosci 10:879–886
Kaur H, Garg A, Raghava GPS (2007) PEPstr: A de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept Lett 14:626–630
Khalili S, Jahangiri A, Borna H, Ahmadi Zanoos K, Amani J (2014) Computational vaccinology and epitope vaccine design by immunoinformatics. Acta Microbiol Immunol Hung 61(3):285–307
Lovell SC, Davis IW, Arendall WB, De Bakker P, Word JM, Prisant MG, Richardson JS, Richardson DC (2002) Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50:437–450
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 16:2785–2791
Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. 2003 Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017
Petersen LR, Roehrig JT (2001) West Nile virus: a reemerging global pathogen. Emerg Infect Dis 7(4):611–614
Petersen LR, Brault AC, Nasci RS (2013) West Nile virus: review of the literature. JAMA 310(3):308–315
Procko E, Gaudet R (2009) Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol 21:84–91
Reche PA, Reinherz EL (2005) PEPVAC: a web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Res 33::W138-42
Ritchie DW, Kozakov D, Vajda S (2008) Accelerating protein–protein docking correlations using a six-dimensional analytic FFT generating function. Bioinform 24:1865–1873
Robinson J, Halliwell JA, Hayhurst JH, Flicek P, Parham P, Marsh SGE (2015) The IPD and IMGT/HLA Database: allele variant databases. Nucleic Acids Res 43:D423-431
Sali WA (2014) Comparative protein structure modeling using modeller. Curr Protoc Bioinform 5(6):1–32
Sharma P, Kumar A (2010) Immunoinformatics: screening of potential T cell antigenic determinants in proteome of H1N1 swine influenza virus for virus epitope vaccine design. J Proteom Bioinform 3:275–278
Sharma P, Saxena K, Mishra S, Kumar A (2014) A comprehensive analysis of predicted HLA binding peptides of JE viral proteins specific to north Indian isolates. Bioinformation 10:334–334
Sharma P, Srivastav S, Mishra S, Kumar A (2017) Sequence and Structure Based Binding Prediction Study of HLA Class I and cTAP Binding Peptides for Japanese Encephalitis Vaccine Development. Int J Pept Res Ther 23:269–279. doi:10.1007/s10989-016-9558-0
Singh SP, Mishra BN (2016) Major histocompatibility complex linked databases and prediction tools for designing vaccines. Hum Immunol 77:295–306
Singh H, Raghava GP (2003) ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics 19(8):1009–1014
Singh A, Mitra M, Sampath G, Venugopal P, Rao JV, Krishnamurthy B, Gupta MK, Sri Krishna S, Sudhakar B, Rao NB, Kaushik Y, Gopinathan K, Hegde NR, Gore MM, Krishna Mohan V, Ella KM (2015a) A Japanese encephalitis vaccine from India induces durable and cross-protective immunity against temporally and spatially wide-ranging global field strains. J Infect Dis 212(5):715–725
Tang H, Liu XS, Fang YZ, Pan L, Zhang ZW et al (2012) The epitopes of foot and mouth disease. Asian J Anim Vet Adv 7:1261–1265
Wallner B, Elofsson A (2003) Can correct protein models be identified. Protein Sci 12:1073–1086
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:407–410
Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V (2005) MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res 33:172–179
Acknowledgements
We are thankful to Prof. A. K. Ghosh, IFTM University, for his experimental guidance during the course of experimental work. We are also grateful to Institute of Biomedical Education and Research, Mangalayatan University, for providing necessary experimental facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research Involving Human and Animal Rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Sharma, P., Sharma, P., Mishra, S. et al. Analysis of Promiscuous T cell Epitopes for Vaccine Development Against West Nile Virus Using Bioinformatics Approaches. Int J Pept Res Ther 24, 377–387 (2018). https://doi.org/10.1007/s10989-017-9624-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-017-9624-2