
Abstract
Cardiomyopathies are a diverse group of cardiac disorders with distinct phenotypes, depending on the proteins and pathways affected. A substantial proportion of cardiomyopathies are inherited and those will be the focus of this review article. With the wide application of high-throughput sequencing in the practice of clinical genetics, the roles of novel genes in cardiomyopathies are recognised. Here, we focus on a subgroup of cardiomyopathy genes [TTN, FHL1, CSRP3, FLNC and PLN, coding for Titin, Four and a Half LIM domain 1, Muscle LIM Protein, Filamin C and Phospholamban, respectively], which, despite their diverse biological functions, all have important signalling functions in the heart, suggesting that disturbances in signalling networks can contribute to cardiomyopathies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Inherited cardiomyopathies (CMs) are genetic diseases of the heart; the majority of them are inherited in an autosomal-dominant (AD) pattern. These diseases can be classified primarily on the basis of dominating morphological and/or functional changes observed in the heart muscle of the affected individual. The different types include dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and left ventricular non-compaction (LVNC) cardiomyopathy (reviewed in Watkins et al. 2011; Yacoub 2014). Many CMs are also associated with life-threatening arrhythmias (reviewed in Bezzina et al. 2015), that potentially result in sudden cardiac death events, making the identification and risk stratification of patients an important issue in the clinical practice.
Despite their different morphological appearances there is significant overlap of the underlying molecular pathways among various CMs. For example, cellular hypertrophy of cardiomyocytes is commonly observed in both DCM and HCM (Davis et al. 2016). Apoptosis, leading to myocyte death, is a prominent feature of DCM and ARVC (Narula et al. 1996; Thiene et al. 1997). Additionally, fibrosis, caused by fibroblast activation and collagen deposition—often in response to apoptosis of cardiomyocytes—is common among DCM, ARVC and HCM (Burlew and Weber 2000).
The genetic aetiology of cardiomyopathies is best understood for HCM, DCM and ARVC. Historically, genomic-wide linkage analysis in large families led to the identification of the first HCM disease gene loci (Solomon et al. 1990; Thierfelder et al. 1993; Watkins et al. 1993). The identification of de novo pathogenic variants in MYH7, the gene encoding sarcomeric beta (β)-Myosin heavy chain, confirmed it as causal gene in HCM (Watkins et al. 1992, 1995). Together with the discovery of pathogenic variants in TPM1 and TNNT2 (encoding the thin filament proteins Tropomyosin and Troponin T) and MYBPC3 (encoding Myosin Binding Protein C), the paradigm of HCM as a “disease of the sarcomere” was postulated (Geisterfer-Lowrance et al. 1990; Thierfelder et al. 1994).
DCM is a more clinically heterogeneous condition. The vast majority of cases have a “non-genetic” aetiology (e.g. ischemic heart disease, viral myocarditis, substance abuse). However, in individuals with so called idiopathic DCM, in whom the common “non-genetic” causes have been excluded, approximately a quarter of cases appear to be familial (Petretta et al. 2011).
Over 50 genes have been reported to cause DCM, either as an isolated phenotype, or as part of a syndrome. These genes encode proteins with a diverse range of structural and functional roles within the cardiac myocyte (e.g. sarcomere, nuclear membrane, desmosome, sarcoplasmic reticulum, cytoskeleton). The majority of non-syndromic DCM is inherited in an AD manner; however autosomal recessive and X-linked forms are also reported (Hershberger et al. 2013; McNally et al. 2013).
In individuals with AD non-syndromic DCM, loss-of-function variants in the TTN gene, which encodes the protein Titin, are the most commonly reported genetic defect (Herman et al. 2012; Pugh et al. 2014; Walsh et al. 2017) and will be discussed below.
Pathogenic variants in LMNA (encoding the nuclear membrane protein isoforms Lamin A and Lamin C), and MYH7 appear to be the second most common, accounting for between 4–6 and 4–5% of cases respectively (Haas et al. 2015; Pugh et al. 2014; Walsh et al. 2017).
Pathogenic variants in many other genes, including protein components of the sarcomere (e.g. TNNT2, TPM1), Z-disk (e.g. TCAP, MYPN, NEXN), cytoskeleton (e.g. DES, VCL), desmosome (e.g. DSP), and RNA-binding proteins (e.g. RBM20), have been reported in DCM cohorts. Individually, these genes appear to account for a smaller proportion of cases (Haas et al. 2015; Pugh et al. 2014; Walsh et al. 2017).
ARVC is recognised as a “disorder of the desmosome”, due to the majority of causal variants arising in genes encoding proteins in this cell–cell contact structure (e.g. PKP2, DSG2, DSC2, DSP and JUP) (Awad et al. 2008).
In the recent years, substantial advances have been made in our understanding of genetic causes of cardiomyopathies through the application of high-throughput genetic sequencing techniques. Genomic sequencing in large reference cohorts has revealed unexpectedly high levels of rare variation in cardiomyopathy genes in the background population (Andreasen et al. 2013; Walsh et al. 2017). Simultaneously, it has become feasible to analyse more candidate genes in larger patient cohorts, and to explore genes which, due to their large size, were technically difficult to analyse (e.g. TTN, DMD and RYR2). This has facilitated the identification of novel disease genes, and enabled re-evaluation of existing gene–disease relationships.
The current major challenge in cardiomyopathy gene analysis is variant interpretation; in many cardiomyopathy disease genes, it is difficult to distinguish between disease-causing and benign variation. Demonstrating the lack of suitable approaches beyond bioinformatics prediction tools, an increasing proportion of variants—especially missense changes—are being classified as “variants of unknown significance” (Alfares et al. 2015; Pugh et al. 2014; Waldmuller et al. 2015; Walsh et al. 2017). Insights into the detailed molecular mechanisms of disease are another challenging aspect of cardiomyopathies and usually lack behind the genetic discoveries.
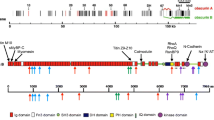
In this review we discuss selected examples of cardiomyopathy genes (TTN, FHL1, CSRP3, FLNC and PLN; see Table 1 and Fig. 1) which, based on their known biological functions and the (limited) functional work on the disease-causing pathogenic variants, have been shown to have important signalling functions in the heart. It is proposed that perturbations of these signalling functions in the presence of pathogenic genetic variants can cause cardiomyopathy.

Schematic localisation of the five proteins of this review in a drawing of a cardiomyocyte; genes name are given in brackets; adapted from Cahill and Gehmlich 2015 with permission
TTN (Titin)
Titin, originally named “connectin” (Maruyama et al. 1977, 1976), appears as a long and flexible filament of about 1 µm in length and 3–4 nm-wide under the electron microscope (Maruyama et al. 1984; Trinick et al. 1984; Wang et al. 1984). Indeed, it is the largest known polypeptide found in nature, a single molecular spans from the Z-disk to M-band of the sarcomere. Titin is encoded by the gene TTN that is located on chromosome 2q31.2. The complete sequence of TTN contains 363 exons, and encodes up to 38,138 amino acid residues with a molecular weight of ~ 4.2 MDa (Bang et al. 2001).
Disease-causing missense pathogenic variants in TTN have been studied extensively in the context of skeletal muscle diseases, including hereditary myopathy with early respiratory failure (HMERF) and tibial muscular dystrophy or Limb Girdle Muscular Dystrophy (LGMD) (Hackman et al. 2002; Pollazzon et al. 2010; Toro et al. 2013). These AD myopathy-causing pathogenic variants are located in C-terminal regions of Titin. Recessive truncating and disruptive missense TTN pathogenic variants have also been linked to disease affecting both skeletal and cardiac muscles, such as early-onset myopathies with fatal cardiomyopathy (Carmignac et al. 2007; Chauveau et al. 2014a, b; Jungbluth and Gautel 2014).
Truncating variants in TTN are the most frequent genetic finding in idiopathic DCM being present in up to 25% of the cases (Herman et al. 2012) and are also found frequently in peri-partum cardiomyopathy (van Spaendonck-Zwarts et al. 2014). This observation was initially confounded by the appearance of TTN truncating variants in normal cohorts (up to 3%) (Roberts et al. 2015), but it is now evident that DCM associated TTN variants tend to cluster predominantly in the A-band portion of Titin, while variants found in controls tend to spare the A-band region and/or are in exons that have low usage in adult cardiac transcripts (Akinrinade et al. 2015a; Roberts et al. 2015; Schafer et al. 2017).
Autosomal dominant TTN missense pathogenic variants have been reported in various types of isolated CM (Gerull et al. 2002; Itoh-Satoh et al. 2002; Matsumoto et al. 2005; Peled et al. 2014; Satoh et al. 1999; Taylor et al. 2011). One of them, TTN W976R, is well supported by co-segregation within a large DCM family and functional data (Gerull et al. 2002; Gramlich et al. 2015; Hinson et al. 2015). Likewise, TTN A178D was identified in a family with features of left-ventricular non-compaction and DCM by a non-biased genetic approach. This pathogenic variant co-segregates with disease in the family and displays impaired function, i.e. reduced binding to the ligand telethonin in vitro (Hastings et al. 2016).
The causality for other TTN missense pathogenic variants is less clear, e.g. next generation sequencing identified large numbers of TTN missense variants in reference populations and in HCM (Lopes et al. 2013), but their relevance for the pathogenesis of HCM remains to be established. The challenge in clinical practice is that missense variants in TTN are common in normal population cohorts. One in three individuals carries a rare (i.e. < 0.01% allelic frequency) variant in TTN, but clearly only a small fraction of these are penetrant disease-causing pathogenic variants. Hence, TTN missense pathogenic variants are currently generally ignored when found in diagnostic sequencing (Akinrinade et al. 2015b; Lopes et al. 2013), however attempts are being made to classify them based on bioinformatics predictions (Haas et al. 2015; Helle and Parikh 2016; Laddach et al. 2017).
Expression of Titin is muscle-specific. Following Myosin and Actin, Titin is the third most abundant protein of striated muscle in vertebrates. As a result of alternative splicing of TTN, a number of Titin isoforms are expressed in different type of striated muscle tissues. In the heart, three major splicing isoforms of Titin have been identified: N2B, N2BA and fetal cardiac Titin. Both N2B and N2BA are isoforms of Titin expressed in adult cardiac muscle (Bang et al. 2001), whereas fetal cardiac Titin is expressed exclusively during development of the fetal heart (Lahmers et al. 2004). N2B is the shortest and stiffest isoform with the size of approximately 3.0 MDa, and is the predominant isoform of Titin expressed in rodent left ventricles (Bang et al. 2001). N2BA is the medium-sized isoform (3.3–3.5 MDa) with compliant stiffness that consists of both N2B and N2A elements, and contains an additional region of PEKV and Immuno-globulin like (Ig) domain elements. The ratio of N2BA to N2B isoforms varies among species and a higher ratio is found in larger animals (Cazorla et al. 2000). In contrast, fetal cardiac Titin is the largest and most compliant cardiac isoform of Titin with a molecular weight of 3.6–3.8 MDa (Lahmers et al. 2004).
A significant increase of N2BA to N2B ratio has been reported in DCM patients, hence affecting the passive tension within the sarcomere due to changes in stiffness (Nagueh et al. 2004). Altered expression ratio between these two Titin isoforms has also been described in a hypertensive myocardium rat model (Warren et al. 2003) and a canine heart failure model (Wu et al. 2002). Of note, RBM20 is an alternative splicing regulator, and pathogenic variants detected in DCM patients have been reported to disrupt protein function. This is associated with more compliant, larger Titin isoforms, which appears to drive the DCM phenotype (Guo et al. 2012). Moreover, this is supported by a RBM20 knockout rat model, which displays a DCM phenotype in the presence of more compliant, larger Titin isoforms.
As an intra-sarcomeric filament, Titin spans a half-sarcomere in length—Titin anchors the Z-disk at its N-terminus, crossing through the I-band and the A-band, towards the M-band at the C-terminus. Titin interacts with different sarcomeric proteins, for instance it binds to α-Actinin (Young et al. 1998), Telethonin (T-cap) (Gregorio et al. 1998; Mues et al. 1998) and may interact with Nebulin’s Src Homology 3 domain (Ma and Wang 2002) at the Z-disk. It binds Actin (Linke et al. 1997) and Obscurin (Young et al. 2001), and interacts with the Myosin filament through Myomesin at the M-line (Fukuzawa et al. 2008; van der Ven and Furst 1997). Cardiac Titin is considered a stable structural and flexible mechanical component of the myocardium, which prevents the overstretching of the sarcomere (Fürst and Gautel 1995; Maruyama 1997; Wang 1984). Titin plays an important role in regulating passive tension, an opposing force created during sarcomere stretching. With the elastic recoil properties largely derived from near the Z-disk towards the edge of the A-band, Titin acts like a “molecular spring” that contributes to the passive tension during cardiac relaxation (Helmes et al. 1999; Linke et al. 1999). The characteristics of this spring can further be modulated by phosphorylation events (reviewed in Hamdani et al. 2017; Kruger and Linke 2011).
In addition, Titin also acts as a scaffold protein for the thick filament proteins within the A-band region (Freiburg and Gautel 1996; Head et al. 2001). In this context, Titin has been suggested to function as a molecular ruler, controlling thick filament length (Bennett and Gautel 1996; Wang 1996; Whiting et al. 1989), however this concept is still controversially discussed (Granzier et al. 2014; Tskhovrebova et al. 2015).
Titin also contains binding sites that interact with several signalling proteins such as protease Calpain p94 (Kinbara et al. 1997), Muscle-specific Ring Finger Protein 1 (MURF-1) (Centner et al. 2001) and Four And A Half LIM Domains 2 (FHL2, also referred to as DRAL) (Lange et al. 2002). In addition, Titin features a serine/threonine kinase domain at the M-line (Gautel et al. 1993). This Titin kinase domain region is conformationally opened by mechanical strain (Puchner et al. 2008), then interacts with Nbr-1 and also recruits MURFs, proteins controlling protein turn-over in cardiomyocytes (Bogomolovas et al. 2014; Lange et al. 2005; Pizon et al. 2002). Moreover, Titin binds FHL1 and FHL2, both of which are implicated in mechano-responsive hypertrophic signalling (Lange et al. 2002; Raskin et al. 2012).
FHL1 (Four And A Half LIM Domains 1)
FHL1 codes for a protein called Four And A Half LIM Domains 1 (FHL1). The gene is positioned on the X-chromosome (Xq26.3), and therefore pathogenic variants in this gene cause X-linked disease. FHL1 was initially identified as a disease gene for skeletal muscle diseases, such as X-linked myopathy with postural muscle atrophy (Windpassinger et al. 2008), reducing body myopathy (Schessl et al. 2009), and Emery–Dreifuss muscular dystrophy (Gueneau et al. 2009). Particularly for Emery–Dreifuss muscular dystrophy cases, cardiac involvement is commonly observed, with conduction defects, arrhythmias, and hypertrophic cardiomyopathy. More recently, FHL1 was also described as a disease gene for HCM, with or without skeletal muscle involvement (Friedrich et al. 2012; Hartmannova et al. 2013; Knoblauch et al. 2010).
FHL1 is a Titin-associated protein, with predominant expression in striated muscle tissues. As the name implies, it consists of four LIM domain and a fold resembling half a LIM domain (Lee et al. 1998). A LIM domain contains a cysteine rich consensus sequence [CX 2 CX 17–19 HX 2 CX 2 CX 2 CX 16–20 CX (2 C/H/D)] and comprises of two zinc fingers which coordinate one zinc ion each (Zheng and Zhao 2007). FHL1 is upregulated in human disease and experimental models of cardiomyopathy (Lu et al. 2012). In particular, the use of an alternative 5′ start site resulting in an “induced” iFHL1 transcript is associated with pathophysiological remodelling (Christodoulou et al. 2014). In the mouse model, inactivation of the gene has no baseline phenotype, however mice lacking FHL1 lack a response to pressure overload in the heart (Sheikh et al. 2008), suggesting that the protein is involved in mechano-signalling pathways. At the molecular level, FHL1 interferes with the phosphorylation of Titin N2B by Extracellular Signal Regulated-Kinase-2 (Erk2), thereby modulating Titin mechanics (Raskin et al. 2012).
Functional work on HCM-causing FHL1 pathogenic variants suggests protein instability and loss of protein as the dominating contributor to disease (Friedrich et al. 2012). Moreover, FHL1 is discussed as a gender-specific modifier of disease severity in HCM patients, given its location on the X chromosome (Christodoulou et al. 2014).
CSRP3 (Muscle LIM Protein)
Muscle LIM Protein (MLP) was initially identified as a regulator of myogenesis in striated muscles (Arber et al. 1994). MLP is encoded by the gene Cysteine and Glycine-rich Protein 3 (CSRP3) on chromosome 11p15.1. Several pathogenic variants in CSRP3 have been shown to cause cardiomyopathies with AD inheritance (Bos et al. 2006; Geier et al. 2003; Hershberger et al. 2008; Mohapatra et al. 2003). Almost all of the reported disease-causing pathogenic variants are located within the first 100 amino acids, no disease-causing variants been identified at the C-terminus (Vafiadaki et al. 2015). The increased availability of next generation sequencing data has helped to validate previously published pathogenic variants. One such variant, CSRP3 p. W4R, described initially as a DCM-causing pathogenic variant (Knoll et al. 2002), has been re-classified as a benign polymorphism (Bos et al. 2006; Geier et al. 2008). Linkage analysis in a large German HCM pedigree led to identification of the C58G missense pathogenic variant in CSRP3 (Geier et al. 2003). The MLP C58G mutant protein, when compared to MLP wildtype, was shown to be more susceptible to degradation in vitro. This supported findings that MLP levels in a cardiac biopsy were significantly reduced, up to 40% in a patient with a heterozygous MLP C58G pathogenic variant (Geier et al. 2008).
In addition to disease-causing pathogenic variants, MLP protein expression changes have been shown to be associated with cardiac disease. MLP was significantly reduced in failing hearts (Zolk et al. 2000), however, as MLP expression is variable in hearts, reduced expression cannot be used as a marker for heart failure.
MLP has been shown to be expressed exclusively in cardiomyocytes and in adult slow-twitch skeletal muscle cells (Arber and Caroni 1996; Schneider et al. 1999). MLP is a relatively small protein, consisting of 194 amino acids. The two LIM domains of MLP are followed by glycine-rich repeat regions, and separated by more than 50 residues. These LIM domains are also responsible for most of the MLP’s protein interactions, both structural and signalling related, in different regions of the cell (Arber and Caroni 1996; Kadrmas and Beckerle 2004; Schmeichel and Beckerle 1994, 1997; Weiskirchen et al. 1995). MLP has been shown to interact with Telethonin (T-cap) (Knoll et al. 2002), α-Actinin (Gehmlich et al. 2004; Louis et al. 1997) and Cofilin-2 (Papalouka et al. 2009) at the Z-disk. In vitro studies have also shown additional binding partners for MLP. For example, MLP can bind to itself (Zolk et al. 2000), it associates with proteins at the costamere (including, Zyxin, Integrin Linked Kinase, and β1-Spectrin) (Flick and Konieczny 2000; Postel et al. 2008; Zolk et al. 2000) and the Nebulin-related Anchoring Protein (N-RAP) (Ehler et al. 2001) at the intercalated disk. MLP also interacts with the nuclear transcription factors MyoD, Myogenin, and Myogenic Regulatory Factor 4 (MRF4) (Kong et al. 1997). MLP’s interactions with these transcription factors, and the presence of predicted nuclear localization signal suggested that MLP function is regulated by translocation between nucleus and cytoplasm (Boateng et al. 2009). There is conflicting information about MLP’s localisation within cardiac cells. MLP has been proposed to be a sarcomere protein located at the Z-disk, I-band, M-line, or at the cell membrane (Arber and Caroni 1996; Arber et al. 1997; Flick and Konieczny 2000; Henderson et al. 2003; Knoll et al. 2010). However, it has also been reported to be a non-sarcomeric protein, with diffuse cytoplasmic expression (Geier et al. 2008).
MLP Knockout (KO) mice were one of the first published models for dilated cardiomyopathy, with a molecular activation of hypertrophic signalling cascades (Arber et al. 1997). MLP KO cardiomyocytes exhibit cytoarchitecture perturbations including disrupted myofibrillar assembly, abnormal alignment of Z-disks and marked fibrosis (Arber et al. 1997). Aberrations at the intercalated discs were observed in these mice, with upregulation of proteins including N-RAP, β-Catenin, Vinculin and plakoglobin, along with upregulation of adherens junctions and downregulation of the gap junction protein Connexin-43 (Ehler et al. 2001). Other studies have also highlighted that loss of MLP leads to perturbation in intracellular calcium handling and excitation–contraction coupling and that a double knockout of MLP and Phospholamban, which regulates sarcoplasmic reticulum calcium intake, rescues the DCM phenotype (Esposito et al. 2000; Kemecsei et al. 2010; Kuhn et al. 2012; Minamisawa et al. 1999, Su et al. 2001).
MLP KO mice are born in Mendelian frequencies, dismissing an indispensable role in embryonic development, however, the protein is thought to be essential for adaptation of the heart to increased hemodynamic stress post birth (Buyandelger et al. 2011). MLP deficiency resulted in loss of passive elasticity in isolated papillary muscles from neonatal and perinatal cardiomyocytes. This has been suggested as a contributing factor to development of diastolic dysfunction and eventual heart failure in these animals. Increased stiffness of cardiomyocytes was also demonstrated by Omens and colleagues in their study performed on hearts from 2-week-old MLP-deficient animals (Omens et al. 2002). The underlying molecular mechanism of this effect, however, is still poorly understood. Prolonged mechanical stress results in maladaptive changes in the cardiomyocytes leading to hypertrophy and eventual heart failure. These observed changes in elasticity, combined with findings that mechanical stimulation failed to stimulate BNP transcription in MLP KO cardiomyocytes, led to the proposal that MLP is part of cardiac stretch sensor complex, along with Titin and Telethonin (Knoll et al. 2002). These suggestions were made considering the findings that MLP was localised to Z-disk. However, more recent findings of MLP’s cytoplasmic localisation (Geier et al. 2008) makes it unlikely that a non-sarcomeric protein such as MLP can be a stress sensor for cardiomyocytes. It is likely that MLP is rather involved in downstream signalling pathways.
MLP heterozygous KO mice (MLP +/−) show no overt phenotype under normal conditions. Compared to WT animals, these mice present with more left ventricular dilation and systolic dysfunction and decreased survival after myocardial infarction; this is associated with a supressed pro-hypertrophic Calcineurin-Nuclear Factor of Activated T-cells (NFAT) signalling pathway (Heineke et al. 2005), again underlining MLP’s role in hypertrophic signalling cascades. Moreover, MLP protein levels have been shown to increase during stress such as aortic banding in wild-type mice (Kuhn et al. 2012). However, overexpression of MLP does not confer any protection to the heart in response to pathological stress such as transverse aortic constriction or chronic infusion of angiotensin-II (Kuhn et al. 2012).
Further, the novel function of MLP as an endogenous inhibitor of Protein Kinase C α (PKCα) in the heart has been elucidated (Lange et al. 2016): Aberrant PKCα signalling in the heart has been shown to cause remodelling and pathological growth of the heart. In the absence of MLP the expression of adapter protein CARP was increased, which led to recruitment of PKCα at the intercalated disc. The absence of CARP reduces PKCα signalling at the intercalated disc, which is why mice lacking both MLP and CARP develop normally and show no signs of DCM (Lange et al. 2016).
FLNC (Filamin C)
Filamin C is encoded by FLNC on chromosome 7q32.1. It is an established disease gene for skeletal muscle disease, causing protein aggregation myofibrillar myopathy (MFM) (Vorgerd et al. 2005) or distal myopathy (Duff et al. 2011). Cardiac involvement has been described for approximately one-third of MFM cases (Kley et al. 2007; Vorgerd et al. 2005). More recently, pathogenic variants in Filamin C were reported in families with familial HCM without skeletal muscle involvement (Valdes-Mas et al. 2014). The majority of the reported putative pathogenic variants were missense changes. In addition, two further missense pathogenic variants were reported in individuals with RCM (Brodehl et al. 2016). Prompted by these findings, screening was expanded onto other types of CMs and subsequently pathogenic variants in FLNC were also associated with DCM and ARVC (Ortiz-Genga et al. 2016). It now emerges that missense pathogenic variants tend to cause HCM or RCM (Brodehl et al. 2016; Gomez et al. 2017), while nonsense and truncation pathogenic variants cause DCM or ARVC (Begay et al. 2016; Janin et al. 2017; Ortiz-Genga et al. 2016).
Filamin C is highly expressed in muscle tissues. It belongs to the family of three Filamin proteins (A, B and C), all characterised by the same modular blueprint (Razinia et al. 2012): at the N-terminus, two calponin- homology domains form an Actin-binding interface, which is followed by 24 Ig-domains. The last of these domains (d24) mediates dimerization of the protein (Himmel et al. 2003; Sjekloca et al. 2007). As a result of this Y-shaped structure, Filamins are Actin-cross linking proteins.
Unique for Filamin C is a striated-muscle specific 80 amino acid long insertion in Ig-domain 20, which mediates interactions to ligands such as e.g. Myotilin (van der Ven et al. 2000), Myopodin (Linnemann et al. 2010), Xin and XIPR2 (van der Ven et al. 2006) and aciculin (Molt et al. 2014). Of note, many of these proteins have striated-muscle specific expression (Myotilin, Myopodin, Xin, XIPR2) and are thought to have a crucial role for the organisation and integrity of skeletal and/or cardiac tissue. For example, Myotilin is a known disease gene for LGMD (Salmikangas et al. 1999) and MFM (Selcen and Engel 2004) and deletion of Xin proteins in mouse models leads to either mild cardiac abnormalities (Otten et al. 2010) or cardiac hypertrophy and electrophysiological changes (Chan et al. 2011; Gustafson-Wagner et al. 2007).
Many of the other Ig-domains have also been found to mediate interactions with ligands (reviewed in van der Flier and Sonnenberg 2001; Zhou et al. 2007) and the protein’s function are modulated by protein phosphorylation events (Murray et al. 2004; Reimann et al. 2017; Sequea et al. 2013).
Like the other members of the Filamin family, Filamin C modulates Actin dynamics. It plays important roles in myofibrillogenesis (Chiang et al. 2000; Dalkilic et al. 2006) by acting in concert with its binding partners Xin, XIRP2 and Aciculin (Molt et al. 2014). A mouse model with genetic inactivation of Filamin C highlights the protein’s crucial role for muscle function; Filamin C deficient mice die at birth due to respiratory failure and have underdeveloped skeletal muscles (Dalkilic et al. 2006).
In mature striated muscle, Filamin C is found at the periphery of the Z-disks, linking sarcomeric Actin structures to the cytoskeleton (Gontier et al. 2005), and at the intercalated disk, a structure which links neighbouring cardiomyocytes to each other. Beyond its structural roles, Filamin C acts as a signalling hub and is an active player in the repair of myofibrillar damage in cardiomyocytes (Leber et al. 2016). Based on its homology and structural similarity with Filamin A, mechano-sensing functions have been postulated (Razinia et al. 2012). The Ig-domains 20–21 of Filamin A have been shown to be in a closed conformation that opens upon mechanical stretch and is subsequently accessible for ligands (Chen et al. 2013; Seppala et al. 2015), thereby providing a molecular basis for how altered mechanical load can trigger downstream signalling events, such as myofibrillar repair.
Filamin C has been identified as a target of chaperone assisted selective autophagy (CASA) (Arndt et al. 2010; Ulbricht et al. 2015). Upon mechanical stress, damaged components of the Z-disk such as Filamin C will be released in a chaperone BAG3-mediated process and targeted for degradation by the autophagosome. This process seems de-regulated in skeletal muscle diseases (especially MFM) when aggregates of mutant Filamin C proteins form (Kley et al. 2013b). These aggregates aberrantly recruit myofibrillar components and hence deplete them from the myofilament (Kley et al. 2013a). Moreover, the CASA mechanism and subsequent autophagy are impaired in the presence of these protein aggregates (Ruparelia et al. 2016).
The patho-mechanisms of Filamin C-related cardiomyopathies are less clear. For DCM, nonsense and truncating pathogenic variants appear to dominate. However, why these pathogenic variants cause pure cardiac disease, mostly without skeletal disease involvement, is still unclear. The absence of Filamin C protein aggregates in the myocardium of DCM patients with Filamin C pathogenic variants is a valuable observation (Ortiz-Genga et al. 2016) and it could be speculated that a loss of function mechanism prevails. In contrast, for some (but not all) HCM/RCM patients with Filamin C pathogenic variants investigated, protein aggregation has been observed in vivo and in vitro (Brodehl et al. 2016; Valdes-Mas et al. 2014). It is currently speculated that depending on the positions of the missense pathogenic variant in the protein, these mutants may cause disease through different modes of action (Gomez et al. 2017).
While Filamin C is now recognised as an important disease gene for cardiomyopathies, future functional work, including the generation of model systems and organisms, is needed to gain insights into the detailed pathophysiology of cardiomyopathies.
PLN (Phospholamban)
Phospholamban is encoded by PLN on chromosome 6q22.31. It is a rare, but well established disease gene for DCM, with several disease-causing missense pathogenic variants identified in familial cohorts. A causative role for PLN R9C in DCM is evidenced by co-segregation in a large 4 generation family affected by DCM and heart failure (Schmitt et al. 2003). Additionally, the L39X pathogenic variant was identified in another large family, resulting in left ventricular hypertrophy in heterozygous carriers and DCM in homozygous carriers in the absence of detectable Phospholamban protein (Haghighi et al. 2003). Interestingly, the heterozygous L39X pathogenic variant has also been found in patients with HCM (Chiu et al. 2007; Landstrom et al. 2011). Another pathogenic variant supported by co-segregation in a large family with DCM is the deletion of arginine 14 (Haghighi et al. 2006), which has also been found in other, unrelated individuals and/or families affected by DCM (DeWitt et al. 2006; Posch et al. 2009). In addition, two pathogenic variants in the promoter region of PLN have been associated with HCM (Medin et al. 2007; Minamisawa et al. 2003), with functional studies showing opposing effects on promotor activity.
Functionally, Phospholamban associates with the Sarcoplasmic Reticulum Calcium ATPase (SERCA2a) (Verboomen et al. 1992) and acts to negatively regulate intracellular calcium removal through direct inhibition of SERCA-mediated calcium uptake into the sarcoplasmic reticulum. Under basal conditions, Phospholamban exists in equilibrium between its monomeric and pentameric form (Fujii et al. 1989), with phosphorylation demonstrated to stabilize the pentameric structure and reduce the affinity of Phospholamban to SERCA2a (Hou et al. 2008). Phosphorylation of serine 16 by Protein Kinase A (PKA) and threonine 17 by Calcium/Calmodulin-dependent Protein Kinase occur in response to beta-adrenergic stimulation (Wegener et al. 1989). Both phosphorylation events release SERCA2a inhibition, thereby increasing SERCA2a’s transport of calcium from the cytosol into the lumen of the sarcoplasmic reticulum during diastole.
Ablation of Phospholamban in mice leads to enhanced myocardial performance (Luo et al. 1994), equivalent to that of wildtype hearts with fully activated by beta-adrenergic stimulation. As such, ablation of Phospholamban has subsequently been used as an experimental approach to improve cardiac function in rodent models of heart failure (Kaneko et al. 2016; Mazzocchi et al. 2016; Minamisawa et al. 1999; Tsuji et al. 2009; Zhang et al. 2010).
Mouse models for DCM-associated PLN pathogenic variants provide sufficient evidence to support a disease-causing role of PLN pathogenic variant in cardiac disease. Transgenic mice carrying the deletion of arginine 14 in Phospholamban die between 2 and 16 weeks of age due to ventricular dilatation and heart failure (Haghighi et al. 2006). At the molecular level, the mutant protein fails to inhibit SERCA2a due to a lack of physical interaction (Haghighi et al. 2012), and instead is mis-localises to the sarcolemmal Na/K-ATPase where it activates its pump function. Transgenic mice expressing the R9C pathogenic variant are also characterised by heart failure and premature death (Schmitt et al. 2003). In these mice, the mutant protein traps PKA and thereby blocks phosphorylation of wildtype Phospholamban. Molecular studies have shown that R9C stabilises the pentameric form of Phospholamban due to disulfide bond formation, preventing phosphorylation by PKA and interaction with SERCA2a (Ha et al. 2011). The R9C transgenic mice have subsequently been used to study disease progression in DCM on the transcriptome and proteome level (Burke et al. 2016; Kuzmanov et al. 2016).
Though pathogenic variants in PLN are rare, findings from PLN mutant carriers and mouse models demonstrate that changes in calcium handling in the presence of Phospholamban pathogenic variants, secondary to perturbations in SERCA2a activity, are sufficient to cause cardiomyopathy.
Conclusions
We have demonstrated with the examples of Titin, FHL1, MLP/Csrp3, Filamin C and Phospholamban discussed here, that there are disease genes for cardiomyopathies beyond the “classical” genes coding for proteins with exclusively structural roles in the sarcomere or the cytoskeleton. It emerges that signalling pathways, often involved in the detection and adaptation to increased load in the normal heart (e.g. acutely upon sympathetic stimulation or more chronically in the presence of hypertension), can be disturbed by pathogenic variants in the genes discussed here and that these chronic disturbances of signalling pathways result in cardiomyopathic changes over a long period of time (often decades).
Our understanding of disease mechanisms lags behind the genetic findings and future work will need to elucidate how pathogenic variants in these genes cause cardiomyopathies. In addition to biochemical in vitro experiments, model organisms such as zebrafish (Asnani and Peterson 2014; Wilkinson et al. 2014) and mice (Camacho et al. 2016) can help gain insight into the complex changes at whole organ level. A novel, emerging technology to model disease in vitro are human induced pluripotent stem cell derived cardiomyocytes, allowing the generation of patient-derived human cardiomyocytes with a specific genetic pathogenic variant. Together with recent advances in genome-editing technologies, induced pluripotent stem cell derived cardiomyocytes have emerged as a powerful tool to explore patho-mechanisms of cardiomyopathies (reviewed in Giacomelli et al. 2017; Sallam et al. 2014).
With exception of TTN truncating variants in DCM, the pathogenic variants in the genes discussed here are individually rare, but collectively they contribute to an estimated 3% of cases in cardiomyopathy cohorts. With the wide-spread application of high-throughput sequencing techniques in the clinical practice, these disease genes will be increasingly interrogated. The challenge remains to confidently assign or disregard a causative role of a variant for the cardiomyopathy phenotype observed in an individual—a classification as “variant of unknown significance” is not helpful e.g. for predictive testing in family members. New bio-informatics approaches in combination with simple, high throughput wet-lab approaches will need to be developed to tackle this challenge.
References
Akininade O, Koskenvuo JW, Alastalo TP (2015a) Prevalence of Titin truncating variants in general population. PLoS One 10:e0145284. https://doi.org/10.1371/journal.pone.0145284
Akinrinade O et al (2015b) Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 36:2327–2337. https://doi.org/10.1093/eurheartj/ehv253
Alfares AA et al (2015) Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 17:880–888. https://doi.org/10.1038/gim.2014.205
Andreasen C et al (2013) New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet 21:918–928. https://doi.org/10.1038/ejhg.2012.283
Arber S, Caroni P (1996) Specificity of single LIM motifs in targeting and LIM/LIM interactions in situ. Genes Dev 10:289–300
Arber S, Halder G, Caroni P (1994) Muscle LIM protein, a novel essential regulator of myogenesis promotes myogenic differentiation. Cell 79:221–231
Arber S et al (1997) MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and. heart failure. Cell 88:393–403
Arndt V et al (2010) Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol 20:143–148. https://doi.org/10.1016/j.cub.2009.11.022
Asnani A, Peterson RT (2014) The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis Model Mech 7:763–767. https://doi.org/10.1242/dmm.016170
Awad MM, Calkins H, Judge DP (2008) Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med 5:258–267. https://doi.org/10.1038/ncpcardio1182
Bang ML et al (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89:1065–1072
Begay RL et al (2016) FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl Sci 1:344–359. https://doi.org/10.1016/j.jacbts.2016.05.004
Bennett PM, Gautel M (1996) Titin domain patterns correlate with the axial disposition of myosin at the end of the thick filament. J Mol Biol 259:896–903. https://doi.org/10.1006/jmbi.1996.0367
Bezzina CR, Lahrouchi N, Priori SG (2015) Genetics of sudden cardiac death. Circ Res 116:1919–1936. https://doi.org/10.1161/CIRCRESAHA.116.304030
Boateng SY, Senyo SE, Qi L, Goldspink PH, Russell B (2009) Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol 47:426–435. https://doi.org/10.1016/j.yjmcc.2009.04.006
Bogomolovas J, Gasch A, Simkovic F, Rigden DJ, Labeit S, Mayans O (2014) Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol 4:140041. https://doi.org/10.1098/rsob.140041
Bos JM et al (2006) Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab 88:78–85. https://doi.org/10.1016/j.ymgme.2005.10.008
Brodehl A et al (2016) Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat 37:269–279. https://doi.org/10.1002/humu.22942
Burke MA et al (2016) Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight. https://doi.org/10.1172/jci.insight.86898
Burlew BS, Weber KT (2000) Connective tissue and the heart functional significance regulatory mechanisms. Cardiol Clin 18:435–442
Buyandelger B, Ng KE, Miocic S, Piotrowska I, Gunkel S, Ku CH, Knoll R (2011) MLP (muscle LIM protein) as a stress sensor in the heart. Pflugers Arch 462:135–142. https://doi.org/10.1007/s00424-011-0961-2
Cahill J, Gehmlich T K (2015) How cardiac cytoarchitecture can go wrong: hypertrophic cardiomyopathy as a paradigm for genetic disease of the heart. In: Ehler E (ed) Cardiac cytoarchitecture: how to maintain a working heart. Springer, Cham, pp 227–243. https://doi.org/10.1007/978-3-319-15263-9_11
Camacho P, Fan H, Liu Z, He JQ (2016) Small mammalian animal models of heart disease. Am J Cardiovasc Dis 6:70–80
Carmignac V et al (2007) C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 61:340–351. https://doi.org/10.1002/ana.21089
Cazorla O et al (2000) Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res 86:59–67
Centner T et al (2001) Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 306:717–726. https://doi.org/10.1006/jmbi.2001.4448
Chan FC et al (2011) Intercalated disc-associated protein, mXin-alpha, influences surface expression of ITO currents in ventricular myocytes. Front Biosci (Elite Ed) 3:1425–1442
Chauveau C et al (2014a) Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet 23:980–991. https://doi.org/10.1093/hmg/ddt494
Chauveau C, Rowell J, Ferreiro A (2014b) A rising titan: TTN review and mutation update. Hum Mutat 35:1046–1059. https://doi.org/10.1002/humu.22611
Chen H, Chandrasekar S, Sheetz MP, Stossel TP, Nakamura F, Yan J (2013) Mechanical perturbation of filamin A immunoglobulin repeats 20–21 reveals potential non-equilibrium mechanochemical partner binding function. Sci Rep 3:1642. https://doi.org/10.1038/srep01642
Chiang W, Greaser ML, Lyons GE (2000) Filamin isogene expression during mouse myogenesis. Dev Dyn 217:99–108. https://doi.org/10.1002/(SICI)1097-0177(200001)217:1<99::AID-DVDY9>3.0.CO;2-5
Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C (2007) Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 43:337–343. https://doi.org/10.1016/j.yjmcc.2007.06.009
Christodoulou DC et al (2014) 5′RNA-Seq identifies Fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest 124:1364–1370. https://doi.org/10.1172/JCI70108
Dalkilic I, Schienda J, Thompson TG, Kunkel LM (2006) Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol Cell Biol 26:6522–6534. https://doi.org/10.1128/MCB.00243-06
Davis J et al (2016) A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 165:1147–1159. https://doi.org/10.1016/j.cell.2016.04.002
DeWitt MM, MacLeod HM, Soliven B, McNally EM (2006) Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol 48:1396–1398. https://doi.org/10.1016/j.jacc.2006.07.016
Duff RM et al (2011) Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet 88:729–740. https://doi.org/10.1016/j.ajhg.2011.04.021
Ehler E et al (2001) Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol 153:763–772
Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ, Rockman HA (2000) Cellular and functional defects in a mouse model of heart failure. Am J Physiol Heart Circ Physiol 279:H3101–H3112
Flick MJ, Konieczny SF (2000) The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. J Cell Sci 113(Pt 9):1553–1564
Freiburg A, Gautel M (1996) A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem 235:317–323
Friedrich FW et al (2012) Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet 21:3237–3254. https://doi.org/10.1093/hmg/dds157
Fujii J, Maruyama K, Tada M, MacLennan DH (1989) Expression and site-specific mutagenesis of phospholamban. Studies of residues involved in phosphorylation and pentamer formation. J Biol Chem 264:12950–12955
Fukuzawa A et al (2008) Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: implications for hereditary myopathies. J Cell Sci 121:1841–1851. https://doi.org/10.1242/jcs.028019
Fürst DO, Gautel M (1995) The anatomy of a molecular giant: how the sarcomere cytoskeleton is assembled from immunoglobulin superfamily molecules. J Mol Cell Cardiol 27:951–959
Gautel M, Leonard K, Labeit S (1993) Phosphorylation of KSP motifs in the C-terminal region of titin in differentiating myoblasts. EMBO J 12:3827–3834
Gehmlich K, Geier C, Osterziel KJ, Van der Ven PF, Furst DO (2004) Decreased interactions of mutant muscle LIM protein (MLP) with N-RAP and alpha-actinin and their implication for hypertrophic cardiomyopathy. Cell Tissue Res 317:129–136. https://doi.org/10.1007/s00441-004-0873-y
Geier C et al (2003) Mutations in the human muscle LIM protein gene in families with. hypertrophic cardiomyopathy. Circulation 107:1390–1395
Geier C et al (2008) Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet 17:2753–2765. https://doi.org/10.1093/hmg/ddn160
Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG (1990) A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62:999–1006
Gerull B et al (2002) Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 30:201–204. https://doi.org/10.1038/ng815
Giacomelli E, Mummery CL, Bellin M (2017) Human heart disease: lessons from human pluripotent stem cell-derived cardiomyocytes. Cell Mol Life Sci. https://doi.org/10.1007/s00018-017-2546-5
Gomez J et al (2017) Screening of the Filamin C gene in a large cohort of hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet. https://doi.org/10.1161/CIRCGENETICS.116.001584
Gontier Y et al (2005) The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J Cell Sci 118:3739–3749. https://doi.org/10.1242/jcs.02484
Gramlich M et al (2015) Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol Med 7:562–576. https://doi.org/10.15252/emmm.201505047
Granzier HL et al (2014) Deleting titin’s I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci USA 111:14589–14594. https://doi.org/10.1073/pnas.1411493111
Gregorio CC et al (1998) The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol 143:1013–1027
Gueneau L et al (2009) Mutations of the FHL1 gene cause Emery–Dreifuss muscular dystrophy. Am J Hum Genet 85:338–353. https://doi.org/10.1016/j.ajhg.2009.07.015
Guo W et al (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18:766–773. https://doi.org/10.1038/nm.2693
Gustafson-Wagner EA et al (2007) Loss of mXinalpha, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol 293:H2680–H2692. https://doi.org/10.1152/ajpheart.00806.2007
Ha KN, Masterson LR, Hou Z, Verardi R, Walsh N, Veglia G, Robia SL (2011) Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc Natl Acad Sci USA 108:2735–2740. https://doi.org/10.1073/pnas.1013987108
Haas J et al (2015) Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 36:1123–1135a. https://doi.org/10.1093/eurheartj/ehu301
Hackman P et al (2002) Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 71:492–500. https://doi.org/10.1086/342380
Haghighi K et al (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111:869–876. https://doi.org/10.1172/JCI17892
Haghighi K et al (2006) A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 103:1388–1393. https://doi.org/10.1073/pnas.0510519103
Haghighi K et al (2012) The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. J Mol Cell Cardiol 52:773–782. https://doi.org/10.1016/j.yjmcc.2011.11.012
Hamdani N, Herwig M, Linke WA (2017) Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys Rev. https://doi.org/10.1007/s12551-017-0263-9
Hartmannova H et al (2013) Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the four-and-a-half LIM domain 1 gene. Circ Cardiovasc Genet 6:543–551. https://doi.org/10.1161/CIRCGENETICS.113.000245
Hastings R et al (2016) Combination of whole genome sequencing, linkage, and functional studies implicates a missense mutation in titin as a cause of autosomal dominant cardiomyopathy with features of left ventricular noncompaction. Circ Cardiovasc Genet 9:426–435. https://doi.org/10.1161/CIRCGENETICS.116.001431
Head JG, Houmeida A, Knight PJ, Clarke AR, Trinick J, Brady RL (2001) Stability and folding rates of domains spanning the large A-band super-repeat of titin. Biophys J 81:1570–1579
Heineke J et al (2005) Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci USA 102:1655–1660. https://doi.org/10.1073/pnas.0405488102
Helle E, Parikh VN (2016) Wrestling the giant: new approaches for assessing titin variant pathogenicity. Circ Cardiovasc Genet 9:392–394. https://doi.org/10.1161/CIRCGENETICS.116.001594
Helmes M, Trombitas K, Centner T, Kellermayer M, Labeit S, Linke WA, Granzier H (1999) Mechanically driven contour-length adjustment in rat cardiac titin’s unique N2B sequence: titin is an adjustable spring. Circ Res 84:1339–1352
Henderson JR, Pomies P, Auffray C, Beckerle MC (2003) ALP and MLP distribution during myofibrillogenesis in cultured cardiomyocytes. Cell Motil Cytoskeleton 54:254–265. https://doi.org/10.1002/cm.10102
Herman DS et al (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366:619–628. https://doi.org/10.1056/NEJMoa1110186
Hershberger RE et al (2008) Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 1:21–26. https://doi.org/10.1111/j.1752-8062.2008.00017.x
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547. https://doi.org/10.1038/nrcardio.2013.105
Himmel M, Van Der Ven PF, Stocklein W, Furst DO (2003) The limits of promiscuity: isoform-specific dimerization of filamins. Biochemistry 42:430–439. https://doi.org/10.1021/bi026501+
Hinson JT et al (2015) HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349:982–986. https://doi.org/10.1126/science.aaa5458
Hou Z, Kelly EM, Robia SL (2008) Phosphomimetic mutations increase phospholamban oligomerization and alter the structure of its regulatory complex. J Biol Chem 283:28996–29003. https://doi.org/10.1074/jbc.M804782200
Itoh-Satoh M et al (2002) Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 291:385–393. https://doi.org/10.1006/bbrc.2002.6448
Janin A et al (2017) Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clin Genet. https://doi.org/10.1111/cge.13043
Jungbluth H, Gautel M (2014) Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci 6:339. https://doi.org/10.3389/fnagi.2014.00339
Kadrmas JL, Beckerle MC (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5:920–931. https://doi.org/10.1038/nrm1499
Kaneko M, Hashikami K, Yamamoto S, Matsumoto H, Nishimoto T (2016) Phospholamban ablation using CRISPR/Cas9 system improves mortality in a murine heart failure. Model PLoS One 11:e0168486. https://doi.org/10.1371/journal.pone.0168486
Kemecsei P et al (2010) Hearts of surviving MLP-KO mice show transient changes of intracellular calcium handling. Mol Cell Biochem 342:251–260. https://doi.org/10.1007/s11010-010-0492-8
Kinbara K, Sorimachi H, Ishiura S, Suzuki K (1997) Muscle-specific calpain, p94, interacts with the extreme C-terminal region of connectin, a unique region flanked by two immunoglobulin C2 motifs. Arch Biochem Biophys 342:99–107. https://doi.org/10.1006/abbi.1997.0108
Kley RA et al (2007) Clinical and morphological phenotype of the filamin myopathy: a study of 31 german patients. Brain 130:3250–3264. https://doi.org/10.1093/brain/awm271
Kley RA et al (2013a) A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol Cell Proteom 12:215–227. https://doi.org/10.1074/mcp.M112.023176
Kley RA, van der Ven PF, Olive M, Hohfeld J, Goldfarb LG, Furst DO, Vorgerd M (2013b) Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy 9:422–423. https://doi.org/10.4161/auto.22921
Knoblauch H et al (2010) Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol 67:136–140. https://doi.org/10.1002/ana.21839
Knoll R et al (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111:943–955
Knoll R et al (2010) A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res 106:695–704. https://doi.org/10.1161/CIRCRESAHA.109.206243
Kong Y, Flick MJ, Kudla AJ, Konieczny SF (1997) Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol 17:4750–4760
Kruger M, Linke WA (2011) The giant protein titin: a regulatory node that integrates myocyte signaling pathways. J Biol Chem 286:9905–9912. https://doi.org/10.1074/jbc.R110.173260
Kuhn C et al (2012) Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res Cardiol 107:262. https://doi.org/10.1007/s00395-012-0262-8
Kuzmanov U et al (2016) Global phosphoproteomic profiling reveals perturbed signaling in a mouse model of dilated cardiomyopathy. Proc Natl Acad Sci USA 113:12592–12597. https://doi.org/10.1073/pnas.1606444113
Laddach A, Gautel M, Fraternali F (2017) TITINdb—a computational tool to assess titin’s role as a disease gene. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx424
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94:505–513. https://doi.org/10.1161/01.RES.0000115522.52554.86
Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ (2011) PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J 161:165–171. https://doi.org/10.1016/j.ahj.2010.08.001
Lange S, Auerbach D, McLoughlin P, Perriard E, Schafer BW, Perriard JC, Ehler E (2002) Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J Cell Sci 115:4925–4936
Lange S et al (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308:1599–1603. https://doi.org/10.1126/science.1110463
Lange S et al (2016) MLP and CARP are linked to chronic PKCalpha signalling in dilated cardiomyopathy. Nat Commun 7:12120. https://doi.org/10.1038/ncomms12120
Leber Y et al (2016) Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum Mol Genet 25:2776–2788. https://doi.org/10.1093/hmg/ddw135
Lee SM et al (1998) Chromosomal mapping, tissue distribution and cDNA sequence of four-and-a-half LIM domain protein 1 (FHL1). Gene 216:163–170
Linke WA, Ivemeyer M, Labeit S, Hinssen H, Ruegg JC, Gautel M (1997) Actin–titin interaction in cardiac myofibrils: probing a physiological role. Biophys J 73:905–919. https://doi.org/10.1016/S0006-3495(97)78123-2
Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC (1999) I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 146:631–644
Linnemann A et al (2010) The sarcomeric Z-disc component myopodin is a multiadapter protein that interacts with filamin and alpha-actinin. Eur J Cell Biol 89:681–692. https://doi.org/10.1016/j.ejcb.2010.04.004
Lopes LR et al (2013) Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet 50:228–239. https://doi.org/10.1136/jmedgenet-2012-101270
Louis HA, Pino JD, Schmeichel KL, Pomies P, Beckerle MC (1997) Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. J Biol Chem 272:27484–27491
Lu B, Yu H, Zwartbol M, Ruifrok WP, van Gilst WH, de Boer RA, Sillje HH (2012) Identification of hypertrophy- and heart failure-associated genes by combining in vitro and in vivo models. Physiol Genom 44:443–454. https://doi.org/10.1152/physiolgenomics.00148.2011
Luo W et al (1994) Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 75:401–409
Ma K, Wang K (2002) Interaction of nebulin SH3 domain with titin PEVK and myopalladin: implications for the signaling and assembly role of titin and nebulin. FEBS Lett 532:273–278
Maruyama K (1997) Connectin/titin, giant elastic protein of muscle. FASEB J 11:341–345
Maruyama K, Natori R, Nonomura Y (1976) New elastic protein from muscle. Nature 262:58–60
Maruyama K, Matsubara S, Natori R, Nonomura Y, Kimura S (1977) Connectin, an elastic protein of muscle characterization function. J Biochem 82:317–337
Maruyama K, Kimura S, Yoshidomi H, Sawada H, Kikuchi M (1984) Molecular size and shape of beta-connectin, an elastic protein of striated muscle. J Biochem 95:1423–1433
Matsumoto Y et al (2005) Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. J Muscle Res Cell Motil 26:367–374. https://doi.org/10.1007/s10974-005-9018-5
Mazzocchi G et al (2016) Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J Physiol 594:3005–3030. https://doi.org/10.1113/JP271622
McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 123:19–26. https://doi.org/10.1172/JCI62862
Medin M, Hermida-Prieto M, Monserrat L, Laredo R, Rodriguez-Rey JC, Fernandez X, Castro-Beiras A (2007) Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN –42 C>G mutation. Eur J Heart Fail 9:37–43. https://doi.org/10.1016/j.ejheart.2006.04.007
Minamisawa S et al (1999) Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99:313–322
Minamisawa S et al (2003) Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun 304:1–4
Mohapatra B et al (2003) Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 80:207–215
Molt S et al (2014) Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J Cell Sci 127:3578–3592. https://doi.org/10.1242/jcs.152157
Mues A, van der Ven PF, Young P, Furst DO, Gautel M (1998) Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett 428:111–114
Murray JT, Campbell DG, Peggie M, Mora A, Cohen P (2004) Identification of filamin C as a new physiological substrate of PKBalpha using. KESTREL Biochem J 384:489–494. https://doi.org/10.1042/BJ20041058
Nagueh SF et al (2004) Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110:155–162. https://doi.org/10.1161/01.CIR.0000135591.37759.AF
Narula J et al (1996) Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335:1182–1189. https://doi.org/10.1056/NEJM199610173351603
Omens JH, Usyk TP, Li Z, McCulloch AD (2002) Muscle LIM protein deficiency leads to alterations in passive ventricular mechanics. Am J Physiol Heart Circ Physiol 282:H680–H687. https://doi.org/10.1152/ajpheart.00773.2001
Ortiz-Genga MF et al (2016) Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 68:2440–2451. https://doi.org/10.1016/j.jacc.2016.09.927
Otten J et al (2010) Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc Res 85:739–750. https://doi.org/10.1093/cvr/cvp345
Papalouka V et al (2009) Muscle LIM protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Mol Cell Biol 29:6046–6058. https://doi.org/10.1128/MCB.00654-09
Peled Y et al (2014) Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol 171:24–30. https://doi.org/10.1016/j.ijcard.2013.11.037
Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D (2011) Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol 108:1171–1176. https://doi.org/10.1016/j.amjcard.2011.06.022
Pizon V et al (2002) Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci 115:4469–4482
Pollazzon M et al (2010) The first Italian family with tibial muscular dystrophy caused by a novel titin mutation. J Neurol 257:575–579. https://doi.org/10.1007/s00415-009-5372-3
Posch MG et al (2009) Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm 6:480–486. https://doi.org/10.1016/j.hrthm.2009.01.016
Postel R, Vakeel P, Topczewski J, Knoll R, Bakkers J (2008) Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin–ECM adhesion complex. Dev Biol 318:92–101. https://doi.org/10.1016/j.ydbio.2008.03.024
Puchner EM et al (2008) Mechanoenzymatics of titin kinase. Proc Natl Acad Sci USA 105:13385–13390. https://doi.org/10.1073/pnas.0805034105
Pugh TJ et al (2014) The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 16:601–608. https://doi.org/10.1038/gim.2013.204
Raskin A et al (2012) A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem 287:29273–29284. https://doi.org/10.1074/jbc.M112.372839
Razinia Z, Makela T, Ylanne J, Calderwood DA (2012) Filamins in mechanosensing and signaling. Annu Rev Biophys 41:227–246. https://doi.org/10.1146/annurev-biophys-050511-102252
Reimann L et al (2017) Myofibrillar Z-discs are a protein phosphorylation hot spot with protein Kinase C (PKCalpha) modulating protein dynamics. Mol Cell Proteom 16:346–367. https://doi.org/10.1074/mcp.M116.065425
Roberts AM et al (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7:270ra276. https://doi.org/10.1126/scitranslmed.3010134
Ruparelia AA, Oorschot V, Ramm G, Bryson-Richardson RJ (2016) FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum Mol Genet 25:2131–2142. https://doi.org/10.1093/hmg/ddw080
Sallam K, Kodo K, Wu JC (2014) Modeling inherited cardiac disorders. Circ J 78:784–794
Salmikangas P, Mykkanen OM, Gronholm M, Heiska L, Kere J, Carpen O (1999) Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum Mol Genet 8:1329–1336
Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A (1999) Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene Biochem. Biophys Res Commun 262:411–417. https://doi.org/10.1006/bbrc.1999.1221
Schafer S et al (2017) Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 49:46–53. https://doi.org/10.1038/ng.3719
Schessl J et al (2009) Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 132:452–464. https://doi.org/10.1093/brain/awn325
Schmeichel KL, Beckerle MC (1994) The LIM domain is a modular protein-binding interface. Cell 79:211–219
Schmeichel KL, Beckerle MC (1997) Molecular dissection of a LIM domain. Mol Biol Cell 8:219–230
Schmitt JP et al (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299:1410–1413. https://doi.org/10.1126/science.1081578
Schneider AG, Sultan KR, Pette D (1999) Muscle LIM protein: expressed in slow muscle and induced in fast muscle by enhanced contractile activity. Am J Physiol 276:C900–C906
Selcen D, Engel AG (2004) Mutations in myotilin cause myofibrillar myopathy. Neurology 62:1363–1371
Seppala J, Tossavainen H, Rodic N, Permi P, Pentikainen U, Ylanne J (2015) Flexible structure of peptide-bound filamin a mechanosensor domain Pair 20–21. PLoS One 10:e0136969. https://doi.org/10.1371/journal.pone.0136969 21
Sequea DA, Sharma N, Arias EB, Cartee GD (2013) Greater filamin C, GSK3alpha, and GSK3beta serine phosphorylation in insulin-stimulated isolated skeletal muscles of calorie restricted 24 month-old rats. Mech Ageing Dev 134:60–63. https://doi.org/10.1016/j.mad.2012.12.002
Sheikh F et al (2008) An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest 118:3870–3880. https://doi.org/10.1172/JCI34472
Sjekloca L et al (2007) Crystal structure of human filamin C domain 23 and small angle scattering model for filamin C 23–24 dimer. J Mol Biol 368:1011–1023. https://doi.org/10.1016/j.jmb.2007.02.018
Solomon SD et al (1990) A locus for familial hypertrophic cardiomyopathy is closely linked to the cardiac myosin heavy chain genes, CRI-L436, and CRI-L329 on chromosome 14 at q11–q12. Am J Hum Genet 47:389–394
Su Z et al (2001) Effects of deletion of muscle LIM protein on myocyte function. Am J Physiol Heart Circ Physiol 280:H2665–H2673
Taylor M et al (2011) Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 124:876–885. https://doi.org/10.1161/CIRCULATIONAHA.110.005405
Thiene G, Basso C, Danieli G, Rampazzo A, Corrado D, Nava A (1997) Arrhythmogenic right ventricular cardiomyopathy a still underrecognized clinic entity. Trends Cardiovasc Med 7:84–90. https://doi.org/10.1016/S1050-1738(97)00011-X
Thierfelder L et al (1993) A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc Natl Acad Sci USA 90:6270–6274
Thierfelder L et al (1994) Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77:701–712
Toro C et al (2013) Exome sequencing identifies titin mutations causing hereditary myopathy with early respiratory failure (HMERF) in families of diverse ethnic origins. BMC Neurol 13:29. https://doi.org/10.1186/1471-2377-13-29
Trinick J, Knight P, Whiting A (1984) Purification and properties of native titin. J Mol Biol 180:331–356
Tskhovrebova L, Bennett P, Gautel M, Trinick J (2015) Titin ruler hypothesis not refuted. Proc Natl Acad Sci USA 112:E1172. https://doi.org/10.1073/pnas.1422326112
Tsuji T et al (2009) Rescue of Ca2+ overload-induced left ventricular dysfunction by targeted ablation of phospholamban. Am J Physiol Heart Circ Physiol 296:H310-317. https://doi.org/10.1152/ajpheart.00975.2008
Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W, Hohfeld J (2015) Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 11:538–546. https://doi.org/10.1080/15548627.2015.1017186
Vafiadaki E, Arvanitis DA, Sanoudou D (2015) Muscle LIM protein: master regulator of cardiac and skeletal muscle functions. Gene 566:1–7. https://doi.org/10.1016/j.gene.2015.04.077
Valdes-Mas R et al (2014) Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun 5:5326. https://doi.org/10.1038/ncomms6326
van Spaendonck-Zwarts KY et al (2014) Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 35:2165–2173. https://doi.org/10.1093/eurheartj/ehu050
van der Ven PF, Furst DO (1997) Assembly of titin, myomesin and M-protein into the sarcomeric M band in differentiating human skeletal muscle cells in vitro. Cell Struct Funct 22:163–171
van der Flier A, Sonnenberg A (2001) Structural and functional aspects of filamins. Biochim Biophys Acta 1538:99–117
van der Ven PF et al (2000) Indications for a novel muscular dystrophy pathway gamma-filamin, the muscle-specific filamin isoform, interacts with myotilin. J Cell Biol 151:235–248
van der Ven PF et al (2006) Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp Cell Res 312:2154–2167. https://doi.org/10.1016/j.yexcr.2006.03.015
Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R (1992) Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem J 286(Pt 2):591–595
Vorgerd M et al (2005) A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet 77:297–304. https://doi.org/10.1086/431959
Waldmuller S et al (2015) Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol Cell Probes 29:308–314. https://doi.org/10.1016/j.mcp.2015.05.004
Walsh R et al (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 19:192–203. https://doi.org/10.1038/gim.2016.90
Wang K (1984) Cytoskeletal matrix in striated muscle: the role of titin, nebulin and intermediate filaments. Adv Exp Med Biol 170:285–305
Wang K (1996) Titin/connectin and nebulin: giant protein rulers of muscle structure and function. Adv Biophys 33:123–134
Wang K, Ramirez-Mitchell R, Palter D (1984) Titin is an extraordinarily long, flexible, and slender myofibrillar protein. Proc Natl Acad Sci USA 81:3685–3689
Warren CM, Jordan MC, Roos KP, Krzesinski PR, Greaser ML (2003) Titin isoform expression in normal and hypertensive myocardium. Cardiovasc Res 59:86–94
Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG, Seidman CE (1992) Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest 90:1666–1671. https://doi.org/10.1172/JCI116038
Watkins H et al (1993) A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet 3:333–337. https://doi.org/10.1038/ng0493-333
Watkins H et al (1995) Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 11:434–437. https://doi.org/10.1038/ng1295-434
Watkins H, Ashrafian H, Redwood C (2011) Inherited cardiomyopathies. N Engl J Med 364:1643–1656. https://doi.org/10.1056/NEJMra0902923
Wegener AD, Simmerman HK, Lindemann JP, Jones LR (1989) Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J Biol Chem 264:11468–11474
Weiskirchen R, Pino JD, Macalma T, Bister K, Beckerle MC (1995) The cysteine-rich protein family of highly related LIM domain proteins. J Biol Chem 270:28946–28954
Whiting A, Wardale J, Trinick J (1989) Does titin regulate the length of muscle thick filaments? J Mol Biol 205:263–268
Wilkinson RN, Jopling C, van Eeden FJ (2014) Zebrafish as a model of cardiac disease. Prog Mol Biol Transl Sci 124:65–91. https://doi.org/10.1016/B978-0-12-386930-2.00004-5
Windpassinger C et al (2008) An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet 82:88–99. https://doi.org/10.1016/j.ajhg.2007.09.004
Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, Granzier H (2002) Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation 106:1384–1389
Yacoub MH (2014) Decade in review—cardiomyopathies: cardiomyopathy on the move. Nat Rev Cardiol 11:628–629. https://doi.org/10.1038/nrcardio.2014.157
Young P, Ferguson C, Banuelos S, Gautel M (1998) Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J 17:1614–1624. https://doi.org/10.1093/emboj/17.6.1614
Young P, Ehler E, Gautel M (2001) Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 154:123–136
Zhang T et al (2010) Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res 106:354–362. https://doi.org/10.1161/CIRCRESAHA.109.207423
Zheng Q, Zhao Y (2007) The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein–protein interaction. Biol Cell 99:489–502. https://doi.org/10.1042/BC20060126
Zhou X, Boren J, Akyurek LM (2007) Filamins in cardiovascular development. Trends Cardiovasc Med 17:222–229. https://doi.org/10.1016/j.tcm.2007.08.001
Zolk O, Caroni P, Bohm M (2000) Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation 101:2674–2677
Acknowledgements
We thank Dr Jillian Simon for comments. KG is supported by a British Heart Foundation Grant (FS/12/40/29712) and acknowledges support from the BHF Centre of Research Excellence, Oxford (RE/13/1/30181). HJ and KG also acknowledge support from the Wellcome Trust (201543/B/16/Z). KLT is the recipient of a National Institute for Health Research (NIHR) doctoral fellowship (NIHR-HCS-D13-04-006). ME is funded by Clarendon Fund doctoral scholarship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ehsan, M., Jiang, H., L.Thomson, K. et al. When signalling goes wrong: pathogenic variants in structural and signalling proteins causing cardiomyopathies. J Muscle Res Cell Motil 38, 303–316 (2017). https://doi.org/10.1007/s10974-017-9487-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-017-9487-3