
Abstract
Treatment of schizophrenia with currently available drugs is often ineffective or results in several adverse reactions. In previous studies focusing on the search for new antipsychotic drugs, we designed and obtained a series of dopamine D2 and serotonin 5-HT1A and 5-HT2A receptor ligands that were pharmacologically evaluated and showed promising antipsychotic activity. Evaluation of ADMET parameters is an important issue in drug development and should be performed at its early stage to avoid developing molecules with poor pharmacokinetics, that are unlikely to enter the market. For this reason, in this work we focused on the assessment of physicochemical parameters of selected compounds from the series we obtained to assess their drug-like potential. The results of thermal analysis showed that most of the tested compounds are thermally stable above 200 °C, with one compound stable up to 190 °C. Permeability through biological membranes assessed in the parallel artificial membrane permeability assay indicated that all tested compounds effectively migrate through biological membranes by means of passive diffusion. The solubility of the tested compounds was determined in PBS, reflecting physiological pH, and 0.01 M HCl, indicating their low to moderate solubility in PBS, which was significantly improved in acidic environment. The lipophilicity of the studied compounds expressed as LogD falls within the range of 1.84–2.80, what suggest that they would show good oral absorption and the ability to cross lipid barriers. The studies were supplemented with in silico prediction of ADMET parameters, which also indicate the probable high drug-likeness of the tested compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antipsychotics are a class of drugs used to treat the symptoms of schizophrenia, which is a mental disorder characterized by an altered perception of reality, resulting from impaired functioning of neurotransmitter circuits in the central nervous system, mainly dopaminergic and glutamatergic [1,2,3]. The exact causes of the disease are still unclear, therefore its current treatment consists in relieving the symptoms. Approved antipsychotics act by either blocking the dopamine D2 receptor (first generation/classical drugs) or, besides that, exerting more potent effect on the serotonin 5-HT2A and 5-HT1A receptors (second and third generation/atypical drugs) [3].
In addition to the desired pharmacodynamic properties, physicochemical parameters are an equally important factor influencing the drug-like potential of newly developed leads. Many drug candidates fail in the late stages of drug development due to poor pharmacokinetics, resulting in a huge financial and time costs [4]. Therefore, it is important to determine the ADMET parameters of potential drug candidates at the initial stages of their development in order to exclude entities with unfavorable properties as early as possible. ADMET refers to pharmacokinetic parameters for drug absorption, distribution, metabolism, excretion and toxicity. These parameters, determined experimentally or computationally predicted, provide valuable information on whether a researched molecule has the potential to become a marketed drug [5].
Another important parameter for pharmaceuticals as well as biologically active compounds is their stability, which can be biological, chemical, and physical. The thermal stability of this type of compounds is closely related to each other. The primary cause of physical instability is the mobility of molecules due to increasing temperatures, while chemical instability is connected with the thermal decomposition of pharmaceutics due to the flow of heat (energy). According to the World Health Organization (WHO), to identify any degradation species in pharmaceutical products, the chemical and thermal stability of products should be evaluated. The thermal stability data of pharmaceuticals provide the required information regarding their handling, storage, shelf life and usage [6,7,8,9,10].
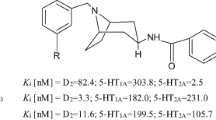
In our previous studies, we attempted to develop new potential antipsychotics by optimizing the virtual hit N-(3-{4-[3-(trifluoromethyl)phenyl]piperazin-1-yl}propyl)-1H-indazole-3-carboxamide (D2AAK3) (Fig. 1) [11]. Thirty-three of its derivatives were synthesized and their affinity for dopamine D2 and serotonin 5-HT1A and 5-HT2A receptors, as the main targets for atypical antipsychotics, was evaluated [12, 13]. Based on the obtained data, selected compounds were subjected to detailed pharmacological and structural studies in order to determine their potential as new drugs in the treatment of schizophrenia.

Chemical structures of D2AAK3 and its derivatives selected for physicochemical evaluation
In this work, our goal was to investigate the physicochemical and drug-like properties of eight most promising compounds (Fig. 1) from the obtained series of D2AAK3 derivatives. The selection of compounds was based on the criteria of atypicality for antipsychotic drugs, i.e., higher affinity for the 5-HT2A receptor than for the D2 receptor. In addition, compounds 1–6 have been identified in previous studies as D2 and 5-HT2A receptor antagonists and 5-HT1A receptor agonists, which is desirable for neuroleptics of newer generations. The affinities of the above compounds for their main targets, determined in our previous work [12, 13], are shown in Table 1.
In order to investigate the thermal stability and determine the temperature-dependent decomposition products of the tested compounds, thermal analyzes were performed using thermogravimetric (TG) and differential scanning calorimetry (DSC) methods. Additionally, the identification of gases released during thermal treatment in oxidizing an inert atmospheres was performed by coupling Fourier-transform infrared (FTIR) spectrophotometer to a thermobalance. The parallel artificial membrane permeability assay (PAMPA) was utilized to assess the permeability of compounds 1–8 through biological membranes. The solubility and lipophilicity of the tested compounds were also experimentally determined, and in silico calculations were performed in order to predict important physicochemical properties from the point of view of ADMET parameters and drug-likeness of potential drugs.
Experimental
Thermal analysis
The thermal behavior of compounds 1–6 in air and nitrogen atmospheres was investigated. The TG, DSC and DTG (derivative thermogravimetry) curves were recorded using Setaram Setsys 16/18 derivatograph between 30 and 700 °C. The samples with masses of 6.58–7.99 mg were heated in an open ceramic crucible with a heating rate 10 °C min−1 in flowing air (v = 0.75 dm3 h−1). The temperature and heat flow of the instrument were calibrated based on the melting point and enthalpy of an indium standard. The thermal analysis with simultaneous registration of gaseous decomposition products was carried out on the TGA Q5000 analyzer (TA Instruments, New Castle, Delaware, USA) connected to the Nicolet 6700 FTIR spectrophotometer (Thermo Scientific). The compounds (15.97–22.24 mg) were heated from 30 to 700 °C in open platinum crucibles under two atmospheres: air and nitrogen (25 mL min−1). The analyses were carried out at a heating rate of 20 °C min−1. The infrared spectra of gaseous products were recorded in the range of 4000–600 cm−1 with a resolution of 4 cm−1 and 6 scans per spectrum. Gas analysis was performed by matching the spectra against those from the spectrum library Nicolet TGA Vapor Phase of the software Ominic and the NIST Chemistry WebBook [14] together with the literature sources.
Permeability profile
Pre-coated PAMPA Plate System Gentest from Corning (Tewksbury, MA, USA) was used in experiments according to manufacturer’s instructions. Briefly, tested compounds and reference drugs were diluted in the PBS buffer (pH 7.4) to the final concentration 100 µM and added to a 96-well donor microplate. Following 5 h of incubation at room temperature the exact quantity of molecules that penetrated from donor to acceptor wells through phospholipid membrane was estimated using the UPLC-MS spectrometry (Waters ACQUITY™ TQD system with the TQ Detector, Waters, Milford, USA). The permeability coefficients (Pe/cm s−1) were calculated using the formula provided by the PAMPA Plate System manufacturer [15].
Solubility and lipophilicity
Equipment and chemicals
UltiMate RS 3000 UHPLC System with Chromeleon 7.2.10 software was purchased from Thermo Fisher Scientific (Waltham, Massachusetts, United States). Cogent Bidentate-C8 chromatographic column, 5 µm, 300 Å, 150 mm × 2.1 mm ID was purchased from Microsolv Technology Corporation (Leland, NC, United States).
Acetonitrile (ACN; for HPLC – super gradient) was purchased from POCH (Gliwice, Poland). Water used in experiments was purified using Milli-Q® Water Purification System from Merck (Darmstadt, Germany). Formic acid LC/MS (~ 98%) and 1-octanol (suitable for HPLC, ≥ 99%) were purchased from SIGMA-ALDRICH (Saint Louis, Missouri, United States). PBS (pH 7.4) and hydrochloric acid (0.1 M analytical solution) were purchased from Chempur (Piekary Śląskie, Poland).
Sample preparation
For solubility determination, about 1 mg of solid sample was precisely weighed (to the nearest 0.01 mg) to glass vial and 4 mL of PBS or 0.01 M HCl, respectively, was added. To prepare standard solution, about 2.5 mg of the solid compound was precisely weighed (to the nearest 0.01 mg) to 25 mL volumetric flask. Then the flask was filled up with water/ACN mixture 1/1 (v/v) to obtain concentration about 100 µg mL−1. All the solutions were shaken for 24 h at 135 rpm using automatic lab shaker. After that, they were left for 1 h without shaking. Next, they were filtered with 0.22 µm syringe filters and used for HPLC analysis.
For LogD determination, about 2.5 mg of the solid compound was precisely weighed (to the nearest 0.01 mg) to 25 mL volumetric flask. 5 mL of 1-octanol was added, then the flask was filled up with water. The solutions were shaken for 24 h at 135 rpm using automatic lab shaker. After that, they were left for 3 h without shaking, to obtain clearly separated liquid phases. A sample of each phase (water and 1-octanol solution) were taken and filtered using 0.22 µm syringe filters. Then, samples were used for HPLC analysis.
HPLC analysis
Mobile phase gradient was obtained by mixing ACN and water, both with addition of 0.1% formic acid (v/v, final concentration). Gradient program was set as follows: from 0 to 1 min—0.5% ACN was maintained, from 1 to 6 min—increase of ACN concentration from 0.5 to 95%, from 6 to 7 min—95% ACN was maintained, from 7 to 9 min—decrease of ACN concentration from 95 to 0.5%, from 9 to 12 min—0.5% ACN was maintained. Mobile phase flow was 0.5 mL min−1. 10 µL of sample was injected using autosampler. Solutes were detected at 254 nm wavelength and were identified by comparison of retention time of the analyzed samples and reference standards.
Solubility of investigated compounds in PBS and 0.01 M HCl was calculated on the basis of solute concentration, determined by comparison of peak areas obtained for specific sample and corresponding standard solution. LogD of compounds was calculated on the basis of direct comparison (ratio) of peak areas obtained for specific sample in octanol phase and water phase.
Prediction of physicochemical parameters in silico
In silico ADMET predictions of the studied compounds were performed in the QikProp module [16] of Schrödinger suite of software, v. 2021-4.
Results and discussion
Thermal analysis
The thermal decomposition data for compounds 1–6 are given in Table 2, whereas TG-DSC and TG curves providing information on their thermal properties are shown in Figs. 2 and 3.

TG, DTG and DSC curves recorded for 1–6 under air atmosphere with heating rate 10 °C min−1

TG curves recorded for 1–6 under air and nitrogen atmosphere with heating rate 20 °C min−1
Compounds 1–6, when heated in an air atmosphere at a rate of 10 °C min−1, show good thermal stability, which is a very important parameter for compounds showing biological activity and considering their potential use as drugs. The first changes due to heating were recorded on the DSC curves as the one, sharp endothermic peaks with maxima at 139–183 °C. These processes are not accompanied by a significant mass change, which suggests that the effects observed on the DSC curves are related to the melting process. Enthalpy values calculated from DSC are 13.52–40.78 kJ mol−1. The shape of the peak indicates that compounds were obtained as pure substances, suitable for pharmaceutical applications. The tested compounds show different melting points, which change in the following order: 3 < 1 < 6 < 4 < 2 ≈ 5. The highest melting temperature is observed for compounds 2 and 5, in which 1H-indazole-3-carboxamide and piperazine units are connected via a four-member chain, and the substituent on the benzene ring (-F or –OCH3) is in the para position. The lack of substituent (1) or the presence of substituent in the meta position (3 and 6) in the aromatic ring connected to the piperazine moiety significantly reduces the melting point of the tested compounds by about 18-45 degrees. The number of carbons in the aliphatic chain has less influence on the melting point. Compound 4, which is an analog of 5, has a three-membered aliphatic linkage and its melting point is only five degrees lower. Further heating of compounds causes their degradation and combustion, which proceeds in two steps (Fig. 2). The exception is compound 5; its decomposition occurs in four stages. The course of thermal decomposition for compounds 1–4 and 6 is similar. The significant mass loss (63.35–69.47%) is observed in the first stage, which begins for these compounds at a similar temperature (249–262 °C). During this step, substituents of the indazole core (i.e., fragments containing benzene and piperazine rings) are probably combusted. The resulting decomposition products are unstable and undergo immediate combustion accompanied by a large exothermic effect (Fig. 2). In the case of compound 5, the decomposition process is more complex. The first mass loss starts at a lower temperature (202 °C) than for the other compounds and is only 3.66%, which can be associated with the breaking of the C=O bond. The next two stages start immediately after the previous ones and result in almost 49% and 16% mass loss (Table 2). The last stage of thermal decomposition is similar to the previous compounds, i.e., it is probably related to the combustion of the indazole moiety and an organic matrix, which is accompanied by a significant exothermic effect.
In order to better understand the processes occurring during the thermal analysis, TG curves (Fig. 3) were recorded with simultaneous analysis of gaseous products in the atmosphere of air and nitrogen (Figs. 4–10, Table 3). Differences in the course of the TG curves of the same compounds in the air from those mentioned earlier (Fig. 2) are mainly related to the mass of the samples taken for analysis, the shape and material of the crucible, as well as the rate of heating.

3D FTIR spectra of the gas product released during thermal decomposition of 1 recorded in: a air atmosphere; b nitrogen atmosphere (heating rate 20 °C min−1)

The FTIR spectrum of volatile products of thermal decomposition of 1 in nitrogen recorded at 368 °C and spectra of possible gaseous products

The FTIR spectrum of volatile products of thermal decomposition of 2 in nitrogen recorded at 513 °C and spectra of possible gaseous products

The FTIR spectrum of volatile products of thermal decomposition of 3 in nitrogen recorded at 455 °C and spectra of possible gaseous products

The FTIR spectrum of volatile products of thermal decomposition of 4 in nitrogen recorded at 358 °C and spectra of possible gaseous products

The FTIR spectrum of volatile products of thermal decomposition of 5 in nitrogen recorded at 415 °C and spectra of possible gaseous products

The FTIR spectrum of volatile products of thermal decomposition of 6 in nitrogen recorded at 360 °C and spectra of possible gaseous products
In the case of compound 1, decomposition in the air at a higher heating rate also occurs in two steps. As can be seen in Fig. 4a, the decomposition process in the oxidizing atmosphere is mainly related to the combustion and release of water molecules (4000–3450 cm−1 and 1950–1300 cm−1), carbon dioxide (2450–2300 cm−1) and the trace of carbon monoxide (2200–2050 cm−1) as well as nitrous oxide (2270–2130 cm–1) [17,18,19,20,21]. Furthermore, vibration bands characteristic of aniline appear on the FTIR spectrum at 320 °C and ammonia above 360 °C. The thermal decomposition process of 1 in nitrogen starts at a higher temperature than in the air and takes place in one step. During the pyrolysis of this compound the variety of released gaseous products increases (Fig. 4b), and the following products are likely to be evolved: water, carbon dioxide, aniline, ammonia, diamines (1,2-diaminoethane/1,3-diaminopropane), acetamide, alkanolamines, benzene and N,N-dimethylaniline (Fig. 5, Table 3). Compound 2, heated at a rate of 20 °C min−1, is thermally stable up to 263 °C and then decomposes with an initial mass loss of 60.55% (see Table 2). Similarly to compound 1, this step is mainly associated with water and carbon dioxide release. At temperatures above 300 °C, bands characteristic of nitrous oxide, carbon monoxide and later aniline, benzene, phenol and ammonia appear. Similar gaseous products, except for aniline and benzene, are released in the next stage (382–481 °C). Additionally, vibrations related to the presence of methane appear in the FTIR spectra. The last step of the process is connected with the combustion of the remaining organic matrix. It is associated with the formation of the following gaseous products: water, carbon dioxide and carbon monoxide. In contrast, the pyrolysis process of the compound is one-stage and involves the evolution of the following gaseous products: water, carbon dioxide, ammonia, p-anisidine, phenol, 1,4-diaminobutane and anisole (Fig. 6).
The thermal decomposition of compound 3 is similar to that discussed previously. In the air, a significant mass change starts at roughly the same temperature, i.e., 256 °C and then the decomposition process occurs in three steps. At the beginning of the first stage, water, carbon dioxide and traces of carbon monoxide and nitrous oxide are evolved. Above 313 °C weak bands characteristic of phenol and ammonia appear in the FTIR spectra. At the end of this stage (420 °C), when the phenol emission decreases, bands characteristic of methane appear (in the range 3175–2900 cm−1 with a characteristic maximum at 3016 cm−1 [22]). Vibrations for these gases are observed in the FTIR spectra up to 620 °C; above this temperature, there are only bands of water, carbon dioxide, nitrous oxide and carbon monoxide. During one-stage pyrolysis the following species in the gas phase can occur: water, carbon dioxide, ammonia, m-anisidine, phenol, 1,4-diaminobutane, anisole, benzene and N,N-dimethylaniline (Fig. 7).
Decomposition of 4 in the air at a higher heating rate occurs in two steps. Almost throughout the process of defragmentation and combustion of the compound on the FITR spectra there are bands characteristic of the following molecules: water, carbon dioxide, carbon monoxide, nitrous oxide, 4-fluoroaniline (> 295 °C), ammonia (> 310 °C), and at the end also methane (> 524 °C). However, above 613 °C, the only gases emitted are water, carbon dioxide, nitrous oxide and carbon monoxide. The pyrolysis process is connected with releasing water, carbon dioxide, carbon monoxide, ammonia, 4-fluoroaniline, diamines (1,2-diaminoethane/1,3-diaminopropane), benzene and fluorobenzene (Fig. 8).
The thermal decomposition of 5 in the air with a heating rate of 20 °C min−1 undergoes in four steps (Fig. 3). The first (190–249 °C) stage and the beginning of the second (below 268 °C) one of the thermal analysis are associated with emission of water and carbon dioxide. Further heating results in the emission of the following gases: 4-fluoroaniline (> 280 °C), carbon monoxide (> 268 °C), ammonia (> 313 °C), nitrous oxide, methane (> 326 °C). The last step of the compound decomposition is related to the release of water, carbon dioxide, carbon monoxide, nitrous oxide and ammonia. The pyrolysis of 5 undergoes in two stages and involves the emission of 4-fluoroaniline, water, carbon dioxide, ammonia, 1,4-diaminobutane and alkanolamines (Fig. 9). In the case of compound 6, decomposition in the air at a higher heating rate also proceeds in two steps. During the first stage, the major mass loss is observed, which is caused by emission of the following gases: carbon dioxide and water, as well as above 300 °C also 3-fluoroaniline, carbon monoxide and nitrous oxide. The same gaseous products are emitted in the second step, but the fluoroaniline vibration band is not observed above 525 °C. As can be seen in Fig. 10, the decomposition process of 6 in nitrogen is mainly related to the release of 3-fluoroaniline, ammonia, water, carbon dioxide, diamines (1,2-diaminoethane/1,3-diaminopropane), benzene and N,N-dimethylaniline.
In summary, the thermal stability of compounds 1–6 (with the heating rate of 20 °C min−1) is higher in an inert atmosphere. During the heating of compounds in synthetic air, the decomposition process is more complicated than in nitrogen. The thermal stability of compounds 1–4 and 6 were higher than 200 °C; in the case of 5 was much lower and amounted to 190 °C. The beginning of the thermal decomposition processes is associated with the release of low molecular mass gases, such as carbon dioxide, carbon monoxide, dinitrogen oxide and water. Based on the chemical structure of compounds (Fig. 1) and the results of thermal behavior for 1–6, it could be proposed that their thermal decomposition is started by the cleavage of carboxamide moiety and C–C bonds of aliphatic chain. Next probably piperazine units with aromatic substituents undergo defragmentation and combustion, as evidenced by the appearance of additional bands characteristic of ammonia and aromatic compounds in the FTIR spectra. At the end of the thermal analysis in an oxidizing atmosphere, the fragment containing the fused rings (i.e., the indazole) is likely to burn. As can be seen in Fig. 3, the thermal decomposition of compounds in nitrogen is less complicated, and for most of the tested compounds, it occurs in one major step. The exceptions are compounds 4 (two steps) and 5 (three steps). However, during pyrolysis, more diverse gaseous products are produced, mainly ammonium, carbon dioxide, carbon monoxide, water, aliphatic and aromatic amines and their derivatives or anisole. The presence of such compounds can be explained by the cleavage of the carboxamide moiety, C–C bonds of the aliphatic chain, and C–N bonds in the piperazine ring and between its ring and the aromatic one. However, the type and diversity of volatile substances emitted during the heating of the studied compounds suggest that their pyrolysis process is also based on the radical mechanism. The radicals formed can react with each other in various ways to form conjugated products that can be observed in the FTIR spectra of the emitted gases or low-volatile compounds that can be condensed into a transfer line as well as being a remnant of the organic matrix.
Permeability profile
PAMPA was utilized to analyze the ability of considered structures to passively diffuse through a lipid-infused artificial membranes [23]. UPLC-MS spectrometry was used to precisely quantify the molecules penetrating from donor to acceptor wells through phospholipid membrane. The results, shown in Table 4, were expressed as permeability coefficient Pe calculated according to previously described formulas [15]. Well-permeable Caffeine (Pe = 9.53 × 10−6 cm s−1) and low-permeable Sulpiride (Pe = 0.0141 × 10−6 cm s−1) were used as references in the study. Based on the obtained results, all tested compounds were identified as effectively migrating through biological membranes by means of passive diffusion.
The ability of a substance to penetrate through lipid membranes constitutes one of the key factors affecting the pharmacokinetics of candidates for drugs. This parameter is of high importance particularly in the case of orally administered drugs, which must exhibit good permeability through biological membranes in order to reach the systemic circulation from the digestive tract. The results obtained in the PAMPA test indicate that compounds 1–8 are characterized by high permeability through lipid membranes, therefore they can be expected to exhibit good oral absorption.
Solubility and lipophilicity
Solubility in phosphate buffered saline (PBS) and 0.01 M HCl, and lipophilicity expressed as LogD for compounds 1–8 were determined experimentally. The obtained data are presented in Table 5.
Solubility is a crucial physicochemical parameter for the absorption of a potential drug. In PBS, which reflects physiological pH, most of the tested compounds show low to moderate solubility, with only compound 5 found to be insoluble. However, the calculated LogS in PBS suggests that the tested compounds, with the exception of compound 5, show sufficient solubility for potential drugs administered orally, assuming the criteria proposed by Jorgensen [24], according to which such leads should have a logS greater than − 5.7. In 0.01 M HCl, in turn, all compounds 1–8 show good solubility at a similar level, which can be easily explained by the basic nature of the compounds and their formation of soluble salts in acidic conditions.
Another important physicochemical property in the context of pharmacokinetics is lipophilicity, which determines whether a potential drug is able to penetrate biological membranes. There are several ways to express lipophilicity, the most commonly used being partition coefficient, LogP, and distribution coefficient, LogD. LogP applies only to neutral forms of the compound, while LogD takes into account its different ionization forms. The assessed lipophilicity expressed as LogD in the range of 1.84–2.80, together with sufficient solubility, suggest that the tested compounds would show good oral absorption and the ability to cross lipid barriers, allowing them to penetrate into the central nervous system. This is in agreement with the data obtained in the PAMPA test.
Prediction of physicochemical parameters in silico
Selected molecular properties and descriptors predicted for compounds 1–8 in the QikProp program [16] are shown in Table 6.
The #stars descriptor value determines the overall drug-likeness of the analyzed compounds based on the comparison of calculated properties with those of known drugs. If any property is outside the 95% range of similar values for known drugs, it is marked with star. Compounds 1–8 all showed zero stars and are therefore classified as highly drug-like according to this parameter. In addition, all analyzed compounds were equally classified as active in the central nervous system, showing a value of 1 on a scale of − 2 to 2. Predicted brain/blood partition coefficients (QPlogBB) range from − 0.696 to − 0.376, suggesting an overall moderate/good distribution of compounds 1–8 from the blood to the brain. Moreover, the predicted oral absorption for all tested compounds is high, on a level of at least about 88% for compound 8.
Moreover, none of the analyzed compounds showed a violation of Lipinski's rule of five. This rule of thumb is commonly used in drug discovery as a standard to determine the drug-likeness of new entities, estimating whether their physicochemical properties are appropriate for an orally administered drug. According to this rule, a drug candidate should fulfill the following criteria: a molecular mass of less than 500 Da, not more than five hydrogen bond donors and ten hydrogen bond acceptors, a calculated octanol–water partition coefficient not exceeding 5. One violation of this rule is allowed for a potential drug candidate administered per os [25].
There are many variants of the above rule assessing the drug-like properties of the lead compound. Jorgensen's rule of three specifies that such a compound should have a solubility expressed as LogS greater than − 5.7, Caco-2 cell permeability greater than 22 nm s−1, and should have less than seven primary metabolites [24]. Of the compounds tested, only compounds 2 and 5 show one violation from this rule as their calculated LogS is less than − 5.7. However, the experimentally determined LogS values indicate that compounds 2 and 5 would also meet the solubility criterion defined by Jorgensen. The remaining compounds meet all the criteria of Jorgensen's rule of three.
Another set of criteria for orally active drugs has been proposed by Veber et al. [26]. According to this rule, compounds with a maximum of ten rotatable bonds or a total of no more than twelve hydrogen bond donors and acceptors, and a polar surface area not exceeding 140 Å2 are likely to display high oral bioavailability. According to Veber's rule, compounds 1–8 are all classified as potentially orally active drugs, as none of them show any violations from these criteria.
Conclusions
The aim of the presented study was to evaluate the physicochemical properties and ADMET parameters of selected compounds, which in previous research showed beneficial properties as potential new antipsychotics. Thermal analysis showed that the tested compounds are thermally stable, which is an important factor for potential drugs. High thermostability avoids storage and processing problems for newly developed drugs. The sharp DSC peaks associated with the melting process indicate that they are crystalline, pure substances. Compared to amorphous substances, these compounds show greater chemical and physical stability. The DSC studies are supported by the TG-FTIR analysis, whose results confirmed that compounds were synthesized as pure, non-hygroscopic substances without any traces of residual solvents. In an oxidizing atmosphere, mainly low-molecular-mass combustion products are formed, while in an inert atmosphere a large variety of gaseous compounds related to the defragmentation of 1–6 is observed. The results of thermal analysis studies are of practical importance, as they will aid in choosing the most promising substances for additional tests, assessing their storage and processing conditions, as well as determining their environmental impact during heating.
Oral absorption is a critical pharmacokinetic parameter, the inadequate values of which often result in the rejection of investigated molecules at an early stage of drug discovery. It is affected by several factors, the most important of which are solubility and lipophilicity. All the tested compounds show sufficient solubility, both in PBS and HCl solution, as determined experimentally. Once dissolved, a drug must cross lipid barriers to reach systemic circulation. At this stage, the key factor is the lipophilicity of the compound. The determined LogD for the tested compounds suggest that they would be able to penetrate biological membranes. These findings are supported by the PAMPA test results, which indicated that all the investigated compounds show high permeability through lipid membranes. This is in agreement with in silico ADMET predictions, which suggest at least 88% absorption rate for the studied compounds. Moreover, they meet the criteria for orally active drugs, defined by set of rules, such as Lipinski's rule of five and Jorgensen's rule of three. Suitable lipophilicity and sufficient penetration through lipid membranes also suggest that the tested compounds would be able to cross the blood–brain barrier and reach the brain, which is crucial for potential antipsychotic drugs. In silico predictions also indicate the sufficient distribution of the analyzed compounds from the blood to the brain, classifying them as likely active in the central nervous system.
Data availability
The data that support the findings of this study are available in the Supplementary Information of this article.
References
Patel KR, Cherian J, Gohil K, Atkinson D. Schizophrenia: overview and treatment options. Pharm Ther. 2014;39:638.
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009. https://doi.org/10.1093/schbul/sbp006.
Stępnicki P, Kondej M, Kaczor AA. Current concepts and treatments of schizophrenia. Mol Basel Switz. 2018. https://doi.org/10.3390/molecules23082087.
Wang J, Urban L. The impact of early ADME profiling on drug discovery and development strategy. Drug Discov World. 2004;5:73–86.
Wishart DS. Improving early drug discovery through ADME modelling. Drugs R D. 2007. https://doi.org/10.2165/00126839-200708060-00003.
Jelić D. Thermal stability of amorphous solid dispersions. Mol Basel Switz. 2021. https://doi.org/10.3390/molecules26010238.
Sovizi MR. Thermal behavior of drugs. J Therm Anal Calorim. 2010. https://doi.org/10.1007/s10973-009-0668-1.
Etxebeste M, Durán A, Sanmartín C, González-Peñas E, Plano D, Lizarraga E. Thermal characterization and stability evaluation of leishmanicidal selenocyanate and diselenide derivatives. J Therm Anal Calorim. 2022. https://doi.org/10.1007/s10973-020-10544-x.
Shamsipur M, Pourmortazavi SM, Beigi AAM, Heydari R, Khatibi M. Thermal stability and decomposition kinetic studies of acyclovir and zidovudine drug compounds. AAPS Pharm Sci Tech. 2013. https://doi.org/10.1208/s12249-012-9916-y.
Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. Geneva: World Health Organization; 2009. pp. 87–130. Report No.: 953. https://www.who.int/publications/i/item/WHO_TRS_953.
Kaczor AA, Targowska-Duda KM, Stępnicki P, Silva AG, Koszła O, Kędzierska E, et al. N-(3-{4-[3-(trifluoromethyl)phenyl]piperazin-1-yl}propyl)-1H-indazole-3-carboxamide (D2AAK3) as a potential antipsychotic: in vitro, in silico and in vivo evaluation of a multi-target ligand. Neurochem Int. 2021. https://doi.org/10.1016/j.neuint.2021.105016.
Stępnicki P, Targowska-Duda KM, Martínez AL, Zięba A, Wronikowska-Denysiuk O, Wróbel MZ, et al. Discovery of novel arylpiperazine-based DA/5-HT modulators as potential antipsychotic agents—design, synthesis, structural studies and pharmacological profiling. Eur J Med Chem. 2023. https://doi.org/10.1016/j.ejmech.2023.115285.
Stępnicki P, Wronikowska-Denysiuk O, Zięba A, Targowska-Duda K, Bartyzel A, Wróbel M, et al. el multi-target ligands of dopamine and serotonin receptors for the treatment of schizophrenia based on indazole and piperazine scaffolds—synthesis, biological activity and structural evaluation. J Enzyme Inhib Med Chem. 2023. https://doi.org/10.1080/14756366.2023.2209828.
Wallace WE. Infrared Spectra. In: Linstrom PJ, Mallard WG, editors. NIST Chem WebBook. Gaithersburg: National Institute of Standards and Technology. pp. 20899.
Chen X, Murawski A, Patel K, Crespi CL, Balimane PV. A novel design of artificial membrane for improving the PAMPA model. Pharm Res. 2008. https://doi.org/10.1007/s11095-007-9517-8.
Schrödinger Release 2021–4: QikProp, Schrödinger, LLC, New York, 2021.
Bartyzel A, Kaczor AA, Głuchowska H, Pitucha M, Wróbel TM, Matosiuk D. Thermal and spectroscopic studies of 2,3,5-trisubstituted and 1,2,3,5-tetrasubstituted indoles as non-competitive antagonists of GluK1/GluK2 receptors. J Therm Anal Calorim. 2018. https://doi.org/10.1007/s10973-018-7146-6.
Makowskaya K, Jargieło P, Worzakowska M, Rogulska M. Thermal properties and the decomposition path of novel UV polymers of terpene-based monomer: citronellyl methacrylate. J Therm Anal Calorim. 2023. https://doi.org/10.1007/s10973-023-12655-7.
Plyler EK, Tidwell ED, Maki AG. Infrared absorption spectrum of nitrous oxide (N2O) from 1830 cm−1 to 2270 cm−1. J Res Natl Bur Stand Sect Phys Chem. 1964. https://doi.org/10.6028/jres.068A.006.
Zhao Y, Wexler AS, Hase F, Pan Y, Mitloehner FM. Detecting nitrous oxide in complex mixtures using FTIR spectroscopy: silage gas. J Environ Prot. 2016. https://doi.org/10.4236/jep.2016.712139.
Petruci JFS, Tütüncü E, Cardoso AA, Mizaikoff B. Real-time and simultaneous monitoring of NO, NO2, and N2O using substrate-integrated hollow waveguides coupled to a compact Fourier transform infrared (FT-IR) spectrometer. Appl Spectrosc. 2019. https://doi.org/10.1177/0003702818801371.
Bartyzel A, Kaczor AA. The formation of a neutral manganese(III) complex containing a tetradentate Schiff base and a ketone—synthesis and characterization. J Coord Chem. 2015. https://doi.org/10.1080/00958972.2015.1073268.
Di L, Kerns E. Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. 2nd ed. Amsterdam: Academic Press; 2016.
Jorgensen WL. Efficient drug lead discovery and optimization. Acc Chem Res. 2009. https://doi.org/10.1021/ar800236t.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169–409X(96)00423–1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv Drug Deliv Rev. 2001; https://doi.org/10.1016/S0169-409X(00)00129-0.
Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002. https://doi.org/10.1021/jm020017n.
Funding
This research was funded by the National Science Center (NCN, Poland) under the OPUS grant 2017/27/B/NZ7/01767 (to A.A.K).
Author information
Authors and Affiliations
Contributions
P.S.: performed thermal analysis and prediction of physicochemical parameters in silico, analyzed the data, wrote the manuscript; R.G.: performed solubility and lipophilicity studies, wrote the manuscript; A. B.: supervised thermal analysis, wrote the manuscript; T. K.: performed permeability studies, wrote the manuscript; K.S.: performed permeability studies T. M. W.: designed solubility and lipophilicity studies; A. A. K.: supervised the study, acquired funding. All authors also read and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stępnicki, P., Gwarda, R.Ł., Bartyzel, A. et al. Thermal behavior and determination of physicochemical properties of novel candidates for antipsychotic drugs. J Therm Anal Calorim (2024). https://doi.org/10.1007/s10973-024-13382-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10973-024-13382-3