
Abstract
For powder metallurgy processing, the sintering stage, i.e. the heat treatment of a the powder compact below the melting point of -at least- the major component, is decisive for establishing microstructure and properties. Thorough studying of the chemical and metallurgical processes occurring during sintering is essential for attaining optimal product properties, and sintering has therefore been the focus of investigations for many decades. Thermoanalytical techniques, at best combined with chemical analysis, enable in-situ characterization of the sintering process from many perspectives. When using these techniques in powder metallurgy, it should be considered that the very large specific surface of a powder compact, compared to a solid metallic body, results in much higher reactivity with the surrounding atmosphere. This atmosphere is on the one hand the “external” one, outside the body in the free space of the furnace, and on the other hand the “internal” one within the pore network of the specimen. This paper shows different examples of how critical information about the sintering process can be described by using thermoanalytical techniques combined with mass spectroscopy: e.g. phase transformations and liquid phase formation in the powdered compact, deoxidation and decarburization reactions, and interstitial redistribution in sintered alloy steels prepared through different alloying techniques.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
“Sintering” is a process that converts a more or less compacted body (or even loose powder) to a material with well-defined mechanical and physical properties, the principal driving force being the reduction of the specific surface and thus the surface energy of the disperse system [1,2,3]. Sintering has been done for many millennia on ceramic materials [4], although it is better known as “firing” in that context. Also for metals, this process is older than commonly assumed. The product obtained from the bloomery furnace, which was the only source for iron and steel until the thirteenth century, consisted in fact of fine iron particles sintered together during the reduction process [5]. Later on, noble metal components were obtained by sintering, such as e.g. the platinum rouble produced in Russia in the early nineteenth century. Modern powder metallurgy started with the tungsten filament for lighting bulbs [6], continued with the invention of the WC–Co hardmetal in 1923—which is still the dominating tool material [7]—and afterwards, in the 1930s, with ferrous precision parts used mostly in the automotive industry [8]. Currently, close to 1 million tons of ferrous powder are consumed annually worldwide for production of precision parts [9].
Both for ceramics and for metals, the sintering process has been studied thoroughly, the work mostly focusing on the mechanisms for densification—which is crucial for ceramics, but also e.g. for hardmetals—and on formation and growth of interparticle contacts [1, 3]. Theories describing the transport phenomena within the ceramic or metallic phase(s) resulting in the above phenomena have been developed, both for solid state and for liquid phase sintering. However, powder compacts also contain a huge specific surface that is the driving force for the sintering processes, but is also responsible for a dramatic increase in the reactivity of the system. This is particularly relevant for metallic systems since metals—with only a few exceptions—are thermodynamically unstable under environmental conditions, the oxide being the stable variant. This is in strong contrast to most ceramics, which are thermodynamically stable in air up to high temperatures. The metals we typically use—steel, aluminium, copper—are stabilized simply because the oxidation reactions are kinetically “frozen” under standard conditions. Only if this kinetic inhibition is lifted—at higher temperatures or in presence of corrosive media—the thermodynamically stable state is established.
This holds for all common metals, but it is of particular relevance for disperse metallic systems, loose powders and also powder compacts, because of their much larger specific surface and thus enhanced reactivity. While solid metallic products, obtained e.g. by classical ingot metallurgy, may be processed in air even at high temperatures—as e.g. done in hot rolling of steel sheet –, powder metallurgy systems must be heat-treated, esp. sintered, in protective atmosphere to avoid catastrophic oxidation [10, 11].
Avoiding oxygen pickup during sintering is however only one item that has to be considered; the second is the removal of oxygen that is already present in the starting powders. All metal powders that have been exposed to air at any time—which is the common situation in industrial practice—are covered by layers of adsorbed oxygen and water, of hydroxides and of oxides. This surface oxygen has to be removed (or at least deactivated) in the early stages of sintering, typically the heating section. Oxide layers inhibit the formation of solid metallic interparticle contacts, the more, the more stable they are, as visible e.g. from hardmetals [12], stainless steels or PM aluminium. For the latter material, removal of the surface oxides by reduction is not possible due to the high thermodynamical stability of the oxides, combined with the low melting point of Al. In this case, penetration of the layers by liquid phase has been the main approach to activate sintering [13,14,15], assisted by the addition of Mg as reducing agent [16].
Another aspect that has to be considered is the much lower surface energy of oxides compared to metals. For sintering this means that the driving force is also lower (which is the reason why for successful sintering of ceramics, typically much finer powders are required than for metals). In metallic systems, conversion from oxidic to metallic particle surfaces increases the driving force for sintering and thus enhances the respective processes. Finally, in the case of liquid phase sintering, deoxidation of the particle surfaces is necessary to ensure good wetting of the solid by the liquid phase and thus promote shrinkage. Poor wetting, in contrast, results in expansion and in highly porous products [17].
For studying the chemical reactions during sintering, thermoanalytical techniques combined with chemical analysis such as mass spectrometry have shown to be highly useful. These techniques were introduced into powder metallurgy by Gert Leitner et al. in the early 1990s, with particular focus on hardmetals [18,19,20]. Also for sintered ferrous materials, studying the deoxidation behaviour proved to be highly helpful [21], also for optimizing the sintering behaviour of new alloy systems. In the following, some examples are described.
Experimental techniques
Various metal powders were used as starting materials in the present work. As base powders, water atomized ferrous grades were used, all supplied by Höganäs AB, Sweden, plain iron ASC.100.29 as well as prealloyed grades Astaloy CrL (Fe–1.5Cr–0.2Mo), CrM (Fe–3Cr–0.5Mo) and CrA (Fe–1.85Cr) being employed (remark: all compositions are given in mass%). As alloy elements, elemental powders such as electrolytic Mn and Cr powders were employed a well as master alloy powders produced through high pressure water atomization (UHPWA; powders courtesy of Atomising systems Ltd., Sheffield, UK). This atomization process results in very fine powders with low oxygen content. As carbon carrier, natural graphite powder Kropfmühl UF4 was used.
The powders were weighed to the desired composition and then blended in a tumbling mixer. Compaction was done in a pressing tool with floating die, die wall lubrication being afforded with Multical sizing fluid. The compacting pressure was 600 MPa if not indicated otherwise. Rectangular compacts were pressed with dimensions 55 mm × 10 mm × ca. 7 mm (Charpy impact test bars ISO 5754). From these compacts smaller specimens were cut if necessary. In part also non-compacted mixes or granulates prepared by crushing compacts were employed for STA measurements.
For the thermoanalytical studies, a simultaneous thermal analyser Netzsch STA-441 Jupiter was used with a sensor which was equipped with W–Re thermocouples to enable operation in H2-containing atmospheres. In parallel, a horizontal pushrod dilatometer Netzsch DIL 402 with Al2O3 measuring system was used. Also this system could be operated in H2. For the first experiments, a similar pushrod dilatometer Bähr 801 was employed that was operated in rotary pump vacuum. In both dilatometers, specimens with dimensions 10 mm × 10 mm × 7 mm cut from the bars were tested as well as full-size impact bars. In the latter case, also mechanical properties such as Charpy impact energy or transverse rupture strength could be measured after the dilatometric run. For evolved gas analysis (EGA), both the STA 449 and the DIL 402 could be coupled to a quadrupole mass spectrometer Netzsch Aeolos through a flexible capillary coupling that was heated to 300 °C during operation, to prevent condensation effects.
Typically, heating and cooling rates were 20 K min−1 for the STA and 10 K min−1 for the dilatometer, the latter one being also representative for industrial sintering. The atmospheres used were either reducing, plain H2 or forming gas N2-10%H2, or inert. In the latter case, Ar was preferred to nitrogen to avoid the interference between N2 and CO in the mass spectrometer. All gases used were of 5.0 quality (> 99.999% purity). After the thermoanalytical runs, the specimens were in part characterized by hot fusion analysis (O, N content) and combustion analysis (carbon). With the dilatometric specimens, also metallographic investigations were done.
Deoxidation behaviour of plain iron-carbon compacts
The first experiments were done with compacts prepared from atomized iron powder and natural graphite. Since carbon is not only an alloy element that increases the strength properties of iron but is also a reducing agent, the experiments were done in inert conditions—vacuum—using the Baehr dilatometer. The temperature intervals in which gas-forming reactions occurred were identified by recording the pressure in the vacuum system, which proved to be a simple but surprisingly effective method [22] (see example in Fig. 1).

Dilatometric and pressure graphs for Fe-1.0%C powder compact. DIL Bähr 801, atomized Fe, compacted at 600 MPa, rotary pump vacuum; heating rate 10 K min−1, Tmax = 1280 °C
Here it is evident that there are 3 major temperature “windows” for gas formation, one at about 400 °C, then a very pronounced peak at about 700 °C and finally a broader one at 900… 1100 °C. It was assumed that the first peak indicated decomposition of hydroxides, and the second—very pronounced one—the carbothermal reduction of the surface oxides. Such reduction occurs more or less simultaneously on all surfaces as soon as the thermodynamic conditions for reduction are fulfilled. The third peak—the broader one—indicates the removal of oxygen from within the Fe powder particles and the oxygen trapped in the pressing contacts, which requires diffusion of oxygen to the surfaces to be removed as gaseous compounds.
Dilatometric runs performed on specimens prepared from different powder fractions corroborated the assignment of peaks 2 and 3. The respective pressure graphs are shown in Fig. 2. Evidently, peak 1 is unaffected by the particle size, while peak 2 decreases in intensity with higher particle sizes—which agrees with the lower specific surface. On the other hand, at increasing particle sizes, peak 3 increases in intensity and is shifted to higher temperatures, which indicates that wider diffusion distances have to be covered by the oxygen atoms to travel to the surface where they can be removed.

Pressure graphs for sintering of Fe-1.0%C prepared from different powder fractions. Atomized iron, compacted at 600 MPa. Compacts 10 × 10 × 6 mm as starting material
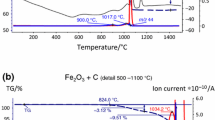
Pressure graphs indicate that gases are generated, i.e. gas-forming reactions take place, which in the present case is a clear indicator for carbothermal reduction processes. However, analytical proof is desirable here. STA-MS runs performed at IKTS Dresden confirmed the assumptions [23]: at T < 500 °C the m18 signal (H2O) dominates (Fig. 3a), indicating in fact desorption of H2O and decomposition of hydroxides. At higher temperatures the m28 signal (CO) is most prominent (Fig. 3b; please note the different scales for the ion current intensity). It is also evident that despite the different methods used—DIL versus STA–, different specimens—compact versus granulate—and the different atmospheres–vacuum versus He—the signals are strikingly similar, just a slight shift of the temperature axis being observed. This shows that at least for a first assessment of the degassing and deoxidation behaviour of powder metallurgy compacts, fairly simple techniques can already be helpful.

Thermochemical (TG-DTG-MS) analysis: Sintering of Fe-1.0%C (water atomized Fe, He-atmosphere-inert). Crushed powder compacts as starting material [23]
The runs described so far have been performed in inert atmosphere (vacuum or He). In industrial practice, however, reducing atmospheres are common, typically N2–H2 mixes, which might result in different deoxidation behaviour. Here it should be considered that, as evident from the Richardson-Ellingham diagrams [24], hydrogen has higher reducing power than carbon at lower temperatures while at high temperatures carbon is more effective. This is a consequence of the increasingly negative Gibbs free energy of CO with higher T, while for H2O—as for all metal oxides—this parameter becomes less negative.
Therefore, STA-MS runs were done with Fe–C both in inert (Ar) and in reducing (H2) atmosphere. To eliminate kinetic effects related to the removal of reaction products, loose powder mixes were used here. The results, given as TG, m18 and m28 graphs, are shown in Fig. 4. As evident, the graphs obtained in Ar are very similar to those presented in Figs. 1 and 3b, with mass losses—and corresponding m28 signals—following the same pattern, just slightly shifted in the temperature axis. In case of the run in H2, in contrast, the pronounced reduction peak that is found at 700… 800 °C under inert conditions is present already at about 400 °C. This reduction is observed in the m18 (H2O) signal, instead of the m28, which agrees with the higher reducing power of H2 in the low temperature range. The m18 peak then however drops—since the surface oxides have been removed—and remains at a fairly low level up to about 850 °C. Above this temperature the m18 signal drops further while the m28 signal strongly increases, indicating that carbon replaces H2 as reducing agent. The broad m28 peak is very similar in shape to that observed in Ar, which means that even in strongly reducing atmospheres, any reduction process that occurs at T > 900 °C will be carbothermal.

Thermoanalytical and chemical (TG-DTG-MS) analysis: Sintering of Fe-0.5%C (water atomized Fe powder, loose powder mix)
For industrial practice this is relevant insofar as carbothermal reduction processes cost carbon, which then is no more available as alloying element. For obtaining a defined “combined carbon content” after sintering—in order to establish the desired mechanical properties—some extra carbon must be added to compensate for the loss involved by reduction. Of course, reproducibility of the carbon loss is an essential precondition for consistent properties of the sintered products. Sintering in H2 or N2-H2 may lower the carbon loss to some extent, but, as is clearly evident from the TG graph in Fig. 4b, the mass loss caused by the carbothermal reduction above 900 °C is about 4 times as high as that linked to reduction with H2. This means that sintering in H2 may lower the carbon loss during sintering by just 20… 25% maximum.
For production of precision parts it must also be considered that the wall thickness of these components may be significantly larger than for test specimens used in thermal analysis, and the diffusion paths from the surface to the core are accordingly longer. For sintering of powder compacts, different atmospheres should be distinguished: the “external” one that surrounds the components, and the “internal” one within the pore network of the part (which at least for pressed, “green”, specimens and in the early stage of sintering is interconnected and open to the surface). This is schematically shown in Fig. 5.

Schematic description of a porous metallic body with “external” and “internal” atmosphere
In case of the external (free) atmosphere, the composition is adjustable by the furnace operator (also with regard to O2, H2O content) and it is affected also by the furnace type, design and gas tightness. Transport of gaseous compounds is performed mainly through convection, in part through diffusion. For the internal atmosphere the composition is mainly determined by local reactions between atmosphere and solid body. Here, the local equilibria are relevant, in particular regarding content of H2O, CO, CO2. Transport of gaseous compounds, in particular of the reaction products, to the surface and into the external atmosphere occurs through diffusion. Convection occurs only through nonisothermal effects, e.g. expansion of the internal atmosphere during heating (“blowing” of the pores) and by generation of gaseous compounds from solids, e.g. carbothermal reduction of oxides.
Since the chemical reactions inside the specimen, esp. the reduction reactions, are controlled by local equilibria, the reactions can only proceed if the reaction products—CO and/or H2O—are transported away, which means that they have to migrate to the surface. I.e. it is the rate at which the reaction products are transported out of the porous body that controls how fast the reaction can proceed inside. Theoretically, also the transport of the reducing agent into the body might play a role, but in case of admixed (or prealloyed) carbon this is irrelevant, since the agent is already inside. Besides, for H2 the diffusion within the pores can be regarded as being much faster than that of the reaction product H2O, i.e. it is the latter that is rate-controlling.
In order to check the effect of kinetic parameters, in particular the transport paths, experiments were carried out with the same material as in Fig. 4, but in this case not loose powder was used but full-size impact test bars, and accordingly the runs were done in the dilatometer. The resulting dilatograms and MS graphs are shown in Fig. 6, both for runs in Ar and in H2, respectively. As can be clearly seen, the pattern is very similar to that shown in Fig. 4, but the peaks are broadened, indicating that the longer diffusion paths in the full-size compacts slow down the reduction reactions. Also the heating of the larger bars can be expected to be slower and less homogeneous, as a consequence of the low thermal conductivity of powder compacts at least in the early stages of sintering [25]. It can therefore be concluded that in larger, esp. thick-walled, specimens the removal of the internal oxides is not quite complete if sintering is done at the standard belt furnace temperature of 1120 °C, because most of the oxygen will be removed at temperatures > 1200 °C, at least for the systems described here. With alloy steels, the situation may be different, as will be described in the following.

Thermoanalytical runs (dilatometer + MS) for Fe-0.5%C. Pressed full size compact; heating rate 10 K min−1, Tmax = 1300 °C, atmosphere argon 5.0 (left)/H2 5.0 (right)
Deoxidation behaviour of alloy steels
Prealloyed grades
In powder metallurgy steels containing metallic alloy elements, the deoxidation behaviour depends mainly on the oxygen affinity of the alloy elements used. For elements such as Cu, Ni and Mo, which form oxides that are similarly or even less stable than iron oxides, the deoxidation behaviour is very similar to that of plain carbon steels, as shown e.g. in [26]. This is exactly the reason why these elements have been traditionally used in sintered steels for many decades. They are however expensive, difficult to recycle and, in case of Ni, toxic. Therefore, cost-effective elements such as Cr, Mn and Si have been introduced to some extent recently. However, the high oxygen affinity poses problems here, both regarding an increased tendency to pick up oxygen from the sintering atmosphere—which can be countered by proper furnace design and improved atmosphere quality—and regarding the more difficult removal of the “natural” oxygen content.
In that context, also the alloying technique plays a major role. In powder metallurgy, more alloying routes are accessible than in ingot metallurgy. There is the prealloyed variant, in which case a suitably alloyed melt is atomized, and each powder particle contains the alloy element(s) in the same concentration. On the other hand, alloy elements can also be admixed as elemental powders to a plain iron base powder, or several alloy elements can be introduced combined through a “masteralloy” powder [27,28,29].
Prealloyed grades available are mostly of Cr or Cr–Mo type since these elements lower the compactibility of a ferrous powder only moderately. Such powders are produced by water atomization with subsequent reducing anneal, and they are covered by a thin iron oxide layer in which small islands of complex (Cr,Mn,Si) oxides are dispersed [30, 31].
In Fig. 7, dilatograms and MS graphs are shown for the higher alloyed variant with 3%Cr. When comparing the MS graphs with those for Fe–C (Fig. 6), it stands out clearly that for the run in Ar the intermediate peak, which is very pronounced for Fe–C, has almost completely disappeared in the Cr steel. However, the high temperature peak is much broader and is in fact a double peak, indicating that in this material removal also of the surface oxides requires much higher temperatures than in case of Fe–C, apparently since these surface oxides are markedly more stable than just the iron oxides. This is at first surprising since the studies done on powders as described in [30, 31] showed the presence of iron oxides which should be more easily reducible.

Thermoanalytical and chemical (TG-DTG-MS) analysis: Sintering of (Fe–3.0%Cr–0.5%Mo)–0.5%C (water atomized Fe powder, compacted at 600 MPa, heating rate 10 K min−1, Tmax = 1300 °C, atmosphere argon 5.0 (left)/H2 5.0 (right)
Sintering in hydrogen atmosphere, in contrast, showed a sharp m18 (H2O) peak at 400 °C, quite similar to that observed for Fe–C, which is a clear indicator that initially, iron oxide is in fact present here. However, as evident from the TG graph, also in this case the fraction of oxygen removed as H2O is insignificant compared to that removed as CO, i.e. carbothermal reduction is the absolutely dominating mechanism, which has to be considered when defining the starting carbon content.
The discrepancy in the low-to-medium temperature reduction behaviour—some reduction with H2, but hardly any with C—was explained by experiments done with compacts presintered in Ar and subsequently full sintered in H2. The m18 peak at 400 °C was taken as the indicator for presence of iron oxide at the surfaces [32]. These experiments showed that already at temperatures below 650 °C the composition of the surface oxides is changed, from easily reducible iron oxides to more stable Cr oxides that require temperatures > 1000 °C for reduction (which is always carbothermal), regardless of the atmosphere used. Furthermore, it is also evident that for reasonably complete oxygen removal, temperatures > 1200 °C are required; therefore, for the Cr and Cr–Mo alloyed steels sintering at 1250 °C or higher is recommended to reap the full potential of these steels regarding the mechanical properties [33,34,35].
Powder mixes
As stated above, the alternative to prealloying is admixing elemental powders. Here it might be assumed that oxygen removal should be uncritical since most of the oxygen introduced into the powder compact is present on the base powder particles, as easily reducible iron oxide. This is however incorrect, as indicated by the graphs in Fig. 8 which correspond to Fe–C compacts to which 4% of elemental Mn was admixed. When comparing these graphs to those shown in Fig. 4 it stands out clearly that for the run in Ar there is surprisingly no medium-temperature reduction peak at 700… 800 °C. There is just only one single big reduction peak at significantly higher temperatures, with its maximum at about 1200 °C, which would rather correspond to carbothermal reduction of Mn oxides than of iron oxides. In case of sintering in H2, there is a small m18 peak at 400 °C, but also here the high temperature m18 peak is absolutely dominating, indicating that highly stable oxides are present.

Thermoanalytical and chemical (TG-DTG-MS) analysis: Sintering of Fe-4.0%Mn-0.5%C (water atomized Fe powder, loose powder mix, heating rate 10 K min−1, Tmax = 1300 °C, atmosphere argon 5.0 (left)/H2 5.0 (right)
The reason for this—at first surprising—behaviour Is the so-called “internal getter” effect [36], which occurs in a powder compact if elements with widely different oxygen affinity are present (as e.g. in case of Fe and Mn). Atmospheres containing reaction products from the carbothermal reduction of iron oxides, i.e. with a fairly high CO content, are strongly oxidizing for Mn (or also for Cr, Si), and therefore any Mn particle will “getter” the oxygen. Such oxygen-sensitive elements will react with CO (or alternatively H2O) to form oxides (e.g. MnO) which are thermodynamically very stable. Carbothermal reduction of such oxides requires high temperatures, as indicated by the m28 peaks shown both in Fig. 8a and b which are in the range above 1100 °C. This “internal getter” effect is schematically shown in Fig. 9.

Schematic description of the “internal getter” effect in powder mixes with heterogeneous oxygen affinity; carbothermal variant
The practical consequence is that although the oxygen originally introduced into the compact is mostly present as easily reducible iron oxides, in fact the sintering conditions should be chosen such as to enable reduction also of the more stable alloy element oxides, which typically means temperatures > 1200 °C. In that respect, there is not much difference if prealloyed or mixed powders are used.
One way to at least alleviate the internal getter effect is the use of masteralloys in which the alloy elements are present with lower chemical activity than in elemental powders; thus their oxygen affinity is lower and also the tendency to getter oxygen from gaseous compounds in the atmosphere [37]. However, the resistance of a masteralloy to internal gettering depends on its composition, as visible from Fig. 10, where TG-MS graphs are shown for Fe-0.5%C with 4% of different masteralloys admixed. Here it can be seen that mixes containing Fe–32Cr–8Si–4C masteralloy show the typical reduction peak for superficial iron oxides between 700 and 800 °C. The intensity of this peak is considerably reduced for the alloy Fe–28Mn–27Cr–3.7C, and the peak completely disappears in mixes containing Fe–42Mn–6Si–0.4C masteralloys, which shows that these latter variants are more prone to internal gettering. Several aspects might influence this behaviour. On one hand the presence of carbon in the masteralloy might be beneficial for preventing oxidation, as well as the presence of passivating elements like Si. On the other hand, Mn, which, because of its high vapour pressure, can sublimate during sintering, might be more readily available as an oxygen getter. Thus, increasing amounts of Mn in the masteralloy might also increase the tendency to act as getter, particularly if the alloy contains only low amounts of C and Si. In any case, most of the oxygen is removed at T > 1000 °C also here, which underlines that for all masteralloy types, sintering at T > 1200 °C is the best way for effective deoxidation.

Thermoanalytical and chemical (TG-MS) analysis: Sintering of Fe-4.0%MA–0.5%C. Water atomized Fe powder, different UHPWA masteralloys, loose powder mix, Ar atmosphere. (a) Masteralloy Fe–32Cr–8Si–4C. (b) Masteralloy Fe–28Mn–27Cr–6Si–3.7C. (c) Masteralloy Fe-42Mn–6S–0.4C
Interaction of gas-forming reactions and densification
In standard pressed and sintered ferrous parts, deoxidation is usually not a problem if the temperature required for reduction of the most stable oxide is attained, since in those materials the pores are interconnected and open to the surface, and they remain so during sintering [38], thus enabling the reduction products to escape from the compact. The situation is however more difficult if systems are involved in which densification occurs during sintering or at least transformation from open to closed pores, since in this case “trapping” of the reduction products will occur, and the reduction comes to a stop.
This is particularly crucial if the temperature “window” for the deoxidation overlaps with that for densification. In case of hardmetals this has been shown e.g. by Gestrich [39]. Normally, for standard WC–Co hardmetals, as well as e.g. for W heavy alloys [40], both “windows” are separated. If however ultrafine hardmetal grades are sintered, densification and resulting pore closure occurs already at fairly low temperatures, as a consequence of the high sintering activity. On the other hand, the deoxidation “window” is shifted to higher temperatures by presence of grain growth inhibitors such as VC or Cr3C2 which form more stable oxides than the base components WC and Co. I.e. both “windows” move towards each other, and if they intersect, pore formation may be the result.
A similar problem may also occur with ferrous PM components, e.g. if the density of the pressed “green” compact is increased to improve the mechanical properties. High pressure compaction [41], warm compaction [42] or high velocity compaction [43] may be such techniques that increase the green density to a level at which the pores are closed already in an early stage of sintering.
One example is shown in Fig. 11: here the degassing (pressure) graphs are given for Cr–Mo steels pressed to two different high density levels by high velocity compaction (HVC) as compared to standard compaction. For this steel grade there is considerable carbothermal reduction in the medium temperature range at all density levels investigated here, but for the lower density material as well as for the medium density grade—the density of which is already rather high for a ferrous PM part—the major reduction peak is observed between 1100 and 1300 °C, as typical for Cr prealloyed steel grades (Fig. 12a, b). The pressure graphs are almost identical, which confirms that the porosity is interconnected and open in both materials, which agrees with the findings in [38]. In case of the very high density material, however, the degassing profile looks quite similar up to a temperature of about 1150 °C; then the reduction peak, which extends to higher temperatures for both other density levels, is cut off (Fig. 12c). This clearly indicates that further reduction has been prevented by pore closing. This is also evident from the as-sintered oxygen content, which is about 5 times higher for the high density material compared to the medium density type. In this case no damage to the microstructure was observed, and the mechanical properties were acceptable, but they were lower than expected regarding the exceptionally high sintered density [34].

Effect of green density on deoxidation of differently compacted steel (Fe-1.5Cr-0.2Mo)-0.5C; dilatometer/vacuum. (a) green density 7.05 g cm−3; oxygen content 0.011%. (b) green density 7.27 g cm−3; oxygen as-sintered 0.026%. (c) green density 7.51 g.cm-3; oxygen as-sintered 0.144%

Sintered steel Fe–Cr–Mo–Mn–Ni–Cu–P–x%C, (1.7%−2.0%−2.5%C), sintered at 1120 °C in N2–H2
This Cr–Mo steel is thus somewhat tolerant to “trapping” of oxygen because it is sintered in solid state, and the starting oxygen content is moderate. In case of liquid phase sintering, in contrast, intersection of the “degassing” and the “densification” windows may render much more spectacular results. In Fig. 12, sections of disk-shaped compacts are shown that were prepared from a complex prealloyed steel powder containing, among others, also Cr and Mn which, as shown above, require fairly high temperatures for reduction of their oxides. Here, the carbon content was varied, which means that while the temperature range for the “deoxidation window” was rather fixed, that for the “densification window” was lowered by higher C levels, which promoted formation of persistent liquid phase. This resulted in the effect that both “windows” progressively intersected, causing pore formation and finally the massive blistering observed for the highest C content. Here, studying the densification and deoxidation behaviour by thermoanalytical techniques combined with chemical analysis is a useful measure to identify such problems.
Conclusions
-
For production of powder metallurgy components, the interactions between the powder compact and the atmosphere during sintering are of utmost importance. This is primarily due to the very large specific surface of a powder compact compared to a solid metallic body.
-
For studying these interactions, thermoanalytical techniques (DTA/DSC/TG; DIL) combined with chemical analysis (EGA) are well suited. However, even simple measures such as recording the pressure in a vacuum system can give a first information.
-
In particular the oxides at the surfaces and in the pressing contacts have to be removed during sintering (which processes may also affect the C content)
-
With traditionally alloyed sintered steels (alloyed with Cu, Ni, Mo), reduction occurs below the sintering temperature, i.e. „is delivered free of charge“ (and is typically not even noticed)
-
Alloy elements with high oxygen affinity (Cr, Mn, Si) require higher temperatures for reduction, in the range of standard sintering temperatures. Selection of the right—not too low—sintering temperature is therefore highly relevant.
-
With compacts containing elements with differing oxygen affinity there is the risk of “internal getter” effects—oxygen transfer from Fe to the alloy element, formation of more stable oxides that are difficult to reduce.
-
Internal gettering occurs with Cr(-Mo) prealloyed sintered steels; the superficial iron oxides are transformed into more stable Cr (Mn, Si, Fe) oxides already at T < 700 °C by diffusion of the alloy element within the particles.
-
In powder mixes. the “Internal getter“ means oxygen transfer from iron oxide to the alloy element particles through the gas phase; this is in fact a metallothermic reduction of Fe oxides with the gas phase as transport path.
-
Less “internal getter“ occurs in mixed systems using masteralloys if the right MA composition is selected, high C and Si contents being helpful here.
-
In practice, most of the O contained is removed at T > 1000 °C anyhow, therefore the safest measure is sintering at T > 1200 °C (this eliminates all internal getter effects)
-
For systems with very high green density or for such that densify during sintering, care must be taken to avoid intersection between the temperature interval for deoxidation and that for pore closing, otherwise at best high residual oxygen contents, at worst pore formation up to massive blistering may occur.
References
Schatt W. Sintervorgänge. Düsseldorf: VDI-Verlag; 1992.
German RM. Sintering theory and practice. New York: J. Wiley & Sons; 1996.
Kang S-JL. Sintering. Oxford: Elsevier-Butterworth Heinemann; 2005.
Wu X, et al. Early pottery at 20,000 years ago in Xianrendong cave, China. Science. 2012;336:1696–700. https://doi.org/10.1126/science.1218643.
Kieffer R, Hotop W. Sintereisen und Sinterstahl. Wien: Springer; 1948.
Johnson PK. Tungsten filaments: the first modern PM product. Int J Powder Metall. 2008;44(4):43–8.
Brookes KJA. Hard metals and other Hard Materials. 2nd ed Int. Carbide Data, UK: East Barnet; 1992.
Whittaker D. Innovation drives Powder Metallurgy structural components forward in the automotive industry. Powder Metall Rev. 2015;4(2):35–53.
Arnhold V, Kruzhanov V. Proc. 39th Hagen symposium 2021 on Powder Metallurgy, in press.
Bradbury S. Powder metallurgy equipment manual, 3rd ed, MPIF, Princeton NJ, 1986.
ASM Handbook Vol.7 Powder metal technologies and applications, ASM, Materials Park OH, 1998 p. 453–67.
Leitner G, Jaenicke-Rößler K, Gestrich T, Breuning T. Shrinkage, liquid phase formation, phase transformation and reactions during the sintering of WC-Co hard metals. Adv Powder Metall & Partic Mater -1997, compiled by R.A.McKotch, R.Webb, MPIF, Princeton NJ 1997; Part 12, p. 75–83.
Storchheim S. Aluminum powder metallurgy finally made commercially practical. Prog Powder Metall. 1962;18:124–30.
Wantanabe T, Yamada K. Effects of methods of adding copper on the strength of sintered aluminium copper alloys. Int J Powder Metall. 1968;4(3):37–47.
Kehl W, Fischmeister HF. Liquid phase sintering of Al-Cu compacts. Powder Metall. 1980;3:113–9. https://doi.org/10.1179/pom.1980.23.3.113.
Kent D, Drennan J, Schaffer GB. A morphological study of nitride formed on Al at low temperature in the presence of Mg. Acta Mat. 2011;59:2469–80. https://doi.org/10.1016/j.actamat.2010.12.050.
Petzow G, Huppmann WJ. Flüssigphasensintern-Verdichtung und Gefügeausbildung. Z Met. 1976;67:579–90.
Leitner G, Heinrich W, Görting K. Thermoanalytical simulation of the sintering behaviour of hard metals. Adv Powder Metall & Partic Mater -1995, compiled by Phillips M, Porter J, MPIF, Princeton NJ 1995; Part 4, p. 259–66.
Leitner G, Hermel W, Jaenicke-Rößler K. In-situ optimization of sintering by gas phase analysis. Adv Powder Metall & Partic Mater -1996, compiled by Cadle TM, Narasimhan KS, MPIF, Princeton NJ 1996; Part 11, p. 435–44.
Gille G, Leitner G, Roebuck B. Sintering behaviour and properties of WC-Co hardmetals in relation to the WC powder properties. Proc EuroPM1995, Birmingham, EPMA, Shrewsbury UK 1995; 195–210.
Gierl-Mayer C. Reactions between ferrous powder compacts and atmospheres during sintering – an overview. Powder Metall. 2020;63(4):237–53. https://doi.org/10.1080/00325899.2020.1810427.
Danninger H, Gerl C, Leitner G, Jaenicke-Rössler K. A simple method to study the degassing and reduction processes during sintering of ferrous powder compacts. PM Sci & Technol Briefs. 2004;6(3):10–4.
Danninger H, Gierl C, Kremel S, Leitner G, Jaenicke-Rössler K, Yu Y. Degassing and deoxidation processes during sintering of unalloyed and alloyed PM steels. Powder Metall Prog. 2002;2(3):125–210.
Glassner AR. The thermochemical properties of the oxides, fluorides, and chlorides to 1500°K. U.S. Atomic Energy Comm Rep. ANL-5750 (1957).
Danninger H, Leitner G, Gierl-Mayer C. Studying the progress of sintering in ferrous powder compacts by in-situ measuring the thermal conductivity. Powder Metall Prog. 2018;18(2):80–95. https://doi.org/10.1515/pmp-2018-0009.
Danninger H, Wolfsgruber E, Ratzi R. Gas formation during sintering of PM steels containing carbon. Proc "EURO PM'97" 1997 Europ Conf on Adv in Structural PM Component Production, Munich, Germany, EPMA, Shrewsbury (1997) 99–106.
Zapf G, Dalal K. Introduction of high oxygen affinity elements manganese, chromium, and vanadium in the powder metallurgy of P/M parts. Mod Dev Powder Metall. 1977;10:129–52.
Banerjee S, et al. New Results in the master alloy concept for high strength sintered steels. Mod Dev Powder Metall. 1981;13:143–57.
De Oro Calderon R, Gierl-Mayer C, Danninger H. Master alloys in powder metallurgy: the challenge of exploring new alloying compositions. Powder Metall. 2017;60:86–96.
Karlsson H, Nyborg L, Berg S, Yu Y. Surface product formation on chromium alloyed steel powder particles. Proc EuroPM2001 Nice Vol.1, EPMA, Shrewsbury (2001) p. 22–7.
Hryha E, Gierl C, Nyborg L, Danninger H, Dudrova E. Surface composition of the steel powders pre-alloyed with manganese. Appl Surf Sci. 2010;256(12):3946–61. https://doi.org/10.1016/j.apsusc.2010.01.055.
Danninger H, de Oro Calderon R, Gierl-Mayer C. Chemical reactions during sintering of PM steel compacts as a function of the alloying route. Powder Metall. 2018. https://doi.org/10.1080/00325899.2018.1458489.
Kremel S, Danninger H, Yu Y. Effect of sintering conditions on particle contacts and mechanical properties of PM steels prepared from 3%Cr prealloyed powder. Powder Metall Prog. 2002;2(4):211–21.
Danninger H, Xu C, Khatibi G, Weiss B, Lindqvist B. Gigacycle fatigue of ultra high density sintered alloy steels. Powder Metall. 2012;55(5):378–87. https://doi.org/10.1179/1743290112Y.0000000001.
Dlapka M, et al. Fatigue behaviour and wear resistance of sinter-hardening steels. Int J Powder Metall. 2012;48(5):49–60.
Gierl-Mayer C, de Oro Calderon R, Danninger H. The role of oxygen transfer in sintering of low alloy steel powder compacts – a review of the “internal getter” effect. JOM. 2016;68(3):920–7. https://doi.org/10.1007/s11837-016-1819-z.
de Oro Calderon R, Gierl-Mayer C, Danninger H. Application of thermal analysis techniques to study the oxidation/reduction phenomena during sintering of steels containing oxygen-sensitive alloying elements. J Therm Anal Calorim. 2017;127:91–105. https://doi.org/10.1007/s10973-016-5508-5.
Dlapka M, Danninger H, Gierl C, Lindqvist B. Defining the pores in PM components. Met Powder Rep. 2010;2:30–3. https://doi.org/10.1016/S0026-0657(10)70093-X.
Gestrich T. In situ Charakterisierung der Vorgänge beim Entbindern, Ausgasen und Sintern von WC-Co-Hartmetallmischungen mittels komplexer thermischer Analyse. PhD-Thesis. Dresden, TU 2005.
Ul Mohsin I, Gierl C, Danninger H. Sintering study of injection molded W-8%Ni-2%Cu compacts from mixed powders by thermoanalytical techniques. Int J Refract Metals Hard Mater. 2011;29:532–7. https://doi.org/10.1016/j.ijrmhm.2011.03.006.
Danninger H, Zengin OZ, Drozda M. High pressure compaction of ferrous PM parts. Met Powder Rep. 1986;11:833–8.
Hoganas Handbook Vol.4 Warm Compaction, Höganäs AB, Höganäs, Sweden (1998).
Skoglund P. High density PM parts by high velocity compaction. Powder Metall. 2001;44(3):199–201.
Funding
Open access funding provided by TU Wien (TUW).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. All authors read and approved the final manuscript.
Corresponding author
Additional information
Dedicated to Gert Leitner, Dresden (1940–2019), a pioneer of thermal analysis in powder metallurgy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Oro Calderon, R., Gierl-Mayer, C. & Danninger, H. Thermoanalytical techniques for characterizing sintering processes in ferrous powder metallurgy. J Therm Anal Calorim 148, 1309–1320 (2023). https://doi.org/10.1007/s10973-022-11740-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-022-11740-7