
Abstract
Graphite, one of the polymorphic forms of carbon, has become a versatile industrial material of nowadays due to its particular attributes. It is used mainly in the automobile industry, metal extractive industry and in the high-tech industry. Moreover, it is also included in the list of critical raw materials for the EU. Our aim was to prove the presence of graphite by thermal analysis beyond X-ray powder diffraction (XRD) and Raman spectroscopy. Thermogravimetry yields comparable results with quantitative XRD. The formation conditions are described by Raman spectrometry and microscopy examinations of drill core samples from Szendrőlád (Szendrő Mts, NE-Hungary; (Szendrőlád Limestone Formation, middle-late Devonian, shelf-basin facies). Polished rock slabs were made for optical microscopy, scanning electron microscopy with energy dispersive spectrometry (SEM–EDS) and Raman spectroscopy. X-ray powder diffraction (XRD) and thermal analysis (DTA-TG) measurements were made on powders. Based on our results, the graphite is epigenetic; its quantity varies between 1.5–3 mass% in the samples. It was developed in 20–50 μm sized flakes, which are often arranged in > 300 μm sized aggregates. Graphite was formed during regional metamorphism from the organic matter-rich shales. The average formation temperature, calculated from the results of Raman spectroscopy, is around 410 °C (± 30 °C). The Raman measurements also indicated the presence of a partially graphitized (disordered graphite) material beside graphite.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Besides diamond and fullerenes, graphite is one of the most common forms of crystalline carbon in nature [1]. It crystallizes with hexagonal or trigonal crystal symmetry, both polytypes (2H and 3R) have layered structure [2]. Due to its particular physical–chemical attributes, it has become a widely used raw material of nowadays [1, 3, 4]. Recent research is in important part focused on investigating new applications of graphite, like the usage in thermal energy storage systems [5, 6]. Being a base raw material in many high-tech applications, it is also included in the list of critical raw materials for the European Union [7], and thus, research on new or less known occurrences is timely. Although for most applications, the petrol coke-based synthetic graphite is used, the demand for natural raw graphite is continuous [8].
There are many ways to indicate the presence and qualify the graphite in the samples, for example, maximum reflectance measurements (%Rmax), X-ray diffraction investigations and transmission electron microscopy (TEM) [9]. However, each of these methods has serious limitations, like grain size (> 5 μm for %Rmax), crystallites size (> 20–30 nm for XRD) or sample preparation for TEM. Additional information can be acquired from scanning electron microscopy (SEM), Fourier-transform infrared spectroscopy (FTIR), differential thermal analysis (DTA) and Raman spectroscopy [10, 11]. For an acceptably accurate graphite survey, the combination of the above methods is a best choice.
In NE-Hungary several graphite-bearing geological formations, mainly black schists, are documented in the Uppony Mts. [12, 13] and [14, 15] or [16] for the Szendrő Mts. Characteristic graphitic samples were recovered and examined in detail from drill cores of the Szendrőlád-6 borehole, from 278–295 m segment (Szendrő Mts, NE-Hungary). Usually, the graphitic schists were investigated with ore microscopy (reflected light mode) and powder XRD methods. Our previous measurements support these two methods as basic identification tools. However, scanning electron microscopy with energy dispersive spectrometry (SEM–EDS) is required to identify the submicrometric textural emplacement of the graphite among rock-forming minerals. Raman spectroscopy [10, 11] and differential thermal analysis (DTA) [17] were also used to characterize graphite and get more information about the formation conditions. Raman spectroscopy can be applied with the scope of inferring the temperature of metamorphism using the geothermometer formula of Beyssac et al. [18]. DTA characteristics are also useful in relative grain size approximation and degree of crystallinity [17]. Additionally, both of these methods allow for the discrimination between inorganic partially (incipiently) graphitized carbon material and different crystalline forms of graphite.
The scope of our work is to establish the precision and reliability of the different analytical methods in graphitic rock analysis, by using thermoanalytical measurements as a fundamental control.
Experimental
Materials
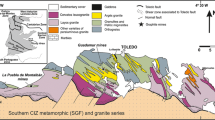
Drill core samples are examined from 278–295 m depth from the Szendrőlád-6 borehole (Fig. 1; Mészégető-valley, Szendrő Mts, NE-Hungary; WGS84 φ = 48° 20′ 39.55521″, WGS84 λ = 20° 43′ 19.38040″, WGS84 h = 265.70 m, depth: 300 m; based on data of MBFSZ [20]). Sample numbering 369 means sample from 278–295 m depth, 371 is from 288 m depth and 373 is from 291 m depth. The graphite bearing rocks are black schists of the Szendrőlád Limestone Formation (middle-late Devonian, shelf-basin facies with epizonal metamorphism of greenschist facies [12], 300–450 °C and 250–300 MPa [21]).

Schematic geological map from the investigated area with the indication of the Szendrőlád-6 borehole (after Gyalog and Síkhegyi [19])
Methods
Polished rock slabs were made for ore microscopy, SEM–EDS and Raman spectroscopy measurements, while bulk powder samples were prepared for XRD and DTA-TG.
For ore microscopy (reflected light mode) examinations a Zeiss Axio.Imager A2.m polarizing petrographic and ore microscope was used with Zeiss AxioCam MRc5 camera. The SEM–EDS analysis was carried out on a JEOL JXA-8600 Superprobe instrument in high vacuum at 20 kV, with beam current of 20 nA (on Faraday cup), with 60 s dwell time for point analysis. Detection limit of the EDS system is 0.1 mass% for light elements and 0.05 mass% for heavy elements, the limit of measurement error is 5 relative%.
For Raman spectroscopy, Thermo Scientific DXR Raman microscope was used with 532 nm wavelength (green) laser, 2 mW laser power, 3 × 15 s exposure time and ~ 4 cm−1 spectral resolution. The spectra were obtained from point measurement of graphite and graphitic flakes in the polished sections. Fityk 1.3.1 software was used for decomposition of Raman spectra with pseudoVoigt function, to extract the individual peaks for band characterization. By Raman spectroscopy, a characteristic spectrum of the graphitic carbonaceous matter can be obtained. For us, the most important is the first-order spectrum, which consists of the well-ordered graphite band at 1580 cm−1 (G band) and the disordered graphite bands at 1350 cm−1 (D1 band), 1620 cm−1 (D2 band) and 1500 cm−1 (D3 band).
Bulk mineralogy on powder samples was measured with X-ray powder diffraction (XRD) (Bruker D8 Advance, Cu K-alfa radiation, 40 kV, 40 mA, parallel beam geometry with Göbel mirror, Vantec-1 PSD detector with 1° opening, 0.007°2θ/24 s counting time). Identification of crystalline phases was done by Search/Match algorithm in DiffracPlus EVA on ICDD PDF2 (2005) database. Rietveld refinement was performed in TOPAS4 software with empirical instrument parameterization on NIST SRM640d Si standard, and crystal structure data for calculations were obtained from AMCSD [22] database. XRD measurements were carried out in the 3DLab Fine Structure Analysis laboratory of the University of Miskolc.
Simultaneous thermal analysis was done by differential thermal analysis, thermogravimetry and derivative thermogravimetry (DTA-TG–DTG), performed on a computer-controlled MOM Derivatograph-C instrument at a linear heating rate of 10 °C min−1 to up to 1000 °C in corundum crucible in air. Samples with similar volume were loaded in the crucible in < 100 mg amount, to assure the total oxidation of carbonaceous material. The TG sensitivity range was set to 100 mg.
Results
Petrography and mineralogy of the samples
The matrix of the samples is built up by V-bearing trioctahedral mica plates (muscovite, muscovite with Na and phengite), embedded in calcite, with largely variable quartz content. Graphite flakes, rarely with low S content, are observed in 20–50 μm size and often arranged in > 300 μm lamellar aggregates (Fig. 2). Graphitic material sometimes also forms aggregates as graphite-TiO2-mica mixtures. All samples have metamorphic texture with intense microfolds, related mainly to the graphitic parts.

a Optical image of graphitic microfolds in calcite matrix, (b) BSE image from the area indicated with white rectangular on figure part (a), (c) EDS spectrum of graphite-muscovite mixture, (d) Element map showing C content of the BSE image on figure part (b)
As accessory minerals, mostly sulfides (pyrite, chalcopyrite, sphalerite, galena, molybdenite and Pb-sulphosalts), phosphates (fluorapatite and monazite), carbonates (siderite, ankerite, dolomite and REE-carbonates – bastnäsite-parisite mixed phases) can be identified beside oxides (Fe-oxide and TiO2 – anatase and rutile, both with Nb and V content) and silicates (zircon, illite, kaolinite, chlorite and rarely albite).
X-ray powder diffraction and Rietveld refinement
XRD results show calcite and muscovite-rich compositions with varying quartz content. Trace amounts of pyrite, rutile, anatase and chlorite are also detected, without direct evidence of graphite due to its low amount, nanocrystalline nature and heavy peak overlapping with quartz peaks (Fig. 3). Graphite peak of (hkl) = (002) (between 26°–27°(2θ)) overlaps with (hkl) = (101) and (hkl) = (110) peaks of quartz. However, deconvolution-based evaluation, like Rietveld refinement allows its direct observation and quantification. The method is based on the modeling of crystalline phases by calculating patterns from the crystal structure to overlap with measured pattern to obtain deconvolute difference curve. Nanocrystalline graphite peaks will be detected on the difference curve, because deconvolute intensities are crystal structure based and overlapping peaks will be resolved. Table 1 shows the detailed quantitative results of the investigated samples calculated by Rietveld refinement. By the deconvolution, a more exact peak position for calcite is made possible and a peak shift for hkl = (104) is observed, indicating Mg (less probably Fe) substitution, theoretical d(104) = 0.3033 nm, while for sample 369, this value is d(104) = 0.30291 nm, for sample 371 is d(104) = 0.30292 nm and for sample 373 d(104) = 0.30286 nm. These peak shifts were fitted together with preferred orientation correction and cation occupancy, resulting in Ca = 0.9 and Mg = 0.1 cation ratios.

XRD patterns of the drill core samples (Ccl—clinochlore, Ms—muscovite, Q—quartz, Cc—calcite, Rt—rutile)
Raman spectroscopy
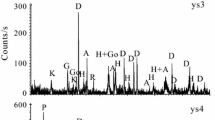
The first-order Raman spectra indicate two sharp bands at ~ 1584 cm−1 (G band) and at ~ 1350 cm−1 (D1 band), representing at the same time the presence of graphite with high degree of atomic ordering and a graphitized material, which did not reach the graphite crystallinity grade [14]. Figure 4 shows one decomposed Raman spectrum indicating the first-order and second-order Raman spectra of graphite beside the peaks of rutile.

Decomposed Raman spectrum of one measurement point
By Raman spectroscopy, the formation temperature (the maximum temperature of metamorphism, without accounting for any existing strain, which would reveal pressure values) can be estimated by the formula of Beyssac et al. [18] through the equation: T (°C) = − 445*R2 + 641 (± 30 °C); where R2 = D1 ⁄ (G + D1 + D2); G = G band area, D1 = D1 band area, D2 = D2 band area.
According to our calculations shown in Table 2, the R2 peak area ratio ranges between 0.43–0.58, corresponding to temperatures from 385 to 448 °C (± 30 °C). In average, the R2 peak area ratio is 0.53, and thus, the formation temperature is ~ 410 °C (± 30 °C) [14].
TG–DTA measurements
According to Földvári [23], if the analysis is performed in air, the exothermic reaction (oxidation) of graphite can occur in a large range, from 600 °C to 1000 °C. As our thermogravimetry analyzes were performed in air, the thermal reaction of graphite can be observed on all curves (Fig. 5). Table 3 (TG) and 4 (DTA) list the reactions and related minerals, in order to delimit graphite and graphitized material oxidation, while Table 5 contains the details of graphite oxidation, DTA peak temperature and associated mass% in the sample. The DTG curves were used in delimiting the different reactions, both for DTA and TG interpretation. Besides graphite, all thermally active minerals identified by XRD-Rietveld refinement are also detected on the simultaneous TG–DTA curves. A number of smaller DTA reactions and TG steps are observed for all samples, which cannot be connected to the phases identified by XRD or SEM–EDS. However, we know that amorphous content of the samples is not totally carbonaceous material, so the early H2O loss (~ 70 °C) and endothermic peaks between 100 and 250 °C can be attributed to X-ray amorphous smectite layers in the mica structure (since no XRD peaks, other than clinochlore, are detected around 0.14 nm), as a result of hydrothermal alteration. This material is an alteration product of silicate minerals for which dehydration indicates smectite type composition, but XRD peaks are not observed due to the small crystallite, size meaning X-ray amorphous nature. Similarly, the reaction in the 500–560 °C region is attributed to illite, present as alteration in the mica structure. The large endothermic preceding calcite decomposition is another OH− loss of mica structure, observed for the very fine grained, “sericite” type appearance of low grade metamorphic or retrograde formations. This peak is overlapping the decomposition of dolomite, observed in minor amount by XRD.

Thermoanalytical curves of the drill core samples
Discussion
Based on our results, the presence of graphite in the samples is proven by most methods independently, as it was identified directly by ore microscopy, Raman spectroscopy and simultaneous TG–DTA measurements. Only indirect identification is possible by Rietveld refinement of XRD data, but it has the advantage of bulk quantification. By SEM–EDS, additional information and petrological-textural attributes are obtained; however, the crystalline nature of carbonaceous material cannot be decided.
According to the calculations of Rietveld refinement, the quantity of graphite varies between 1.5–3 mass% in the samples. In all cases, 3R polytype of graphite was detected by XRD. The polytype (3R or 2H) development depends on the formation pressure, in the case of 3R polytype, hydrostatic pressure-dominated conditions could facilitate its crystallization [17]. In our samples, this is in accordance with the observed accessory minerals, as sulfides and carbonates are indicating a low temperature (below ~ 500 °C) hydrothermal process. From the minor extent of transformations and small amount of developed minerals, the process happened with limited fluid recharge, but which possibly influenced the graphite crystallization, as well. Such a hydrothermal alteration is in accordance also with the smectite, vermiculite traces, as well as illite and “sericite” occurrence, low temperature hydration may produce such phases. Composition and thermal behavior of calcite is signaling higher than normal crystallization temperatures, according to its Mg-content. Theoretically, calcite has an endothermic decomposition with reaction peak at 936 °C if the calcite is well crystallized, its structure is not damaged by weathering and contains no substitution [23]. By Rietveld refinement and SEM–EDS, Mg-content of calcite was evidenced in the samples and accordingly the theoretical calcite endothermic reaction shifted to lower temperature values (~ 830 °C) on the simultaneous TG–DTA curves (Fig. 5 and Table 4). However, due to the “sericite” dehydroxylation and dolomite decomposition estimation of Mg-content is not possible.
By Raman spectroscopy, besides graphite, the presence of a graphitized material was also indicated, but this did not reach the graphite crystallinity grade, termed “disordered graphite” [14]. For such materials, the application of thermogravimetry is useful in tracing the natural organic, semi-organic matter, as there is a correlation between the temperature of exothermal effects during the combustion of the organic matter and the grade of maturation [23]. On the simultaneous TG–DTA curves, the partially graphitized material also could be detected, Table 6 contains the details of the oxidation, DTA peak temperature and associated mass%. In summary, the well-crystallized graphite is associated with carbonaceous material, below the graphite crystallinity grade by Raman spectroscopy, the same material which is partially graphitized according to DTA-TG. Such a material could be X-ray amorphous, meaning such small graphite layers, if any, that diffraction is not detected, thus accounted for in the amorphous fraction also by our measurements. From the results of XRD and thermoanalytical measurements, (Table 7) the graphite, graphitized material and silicate amorphous contents of the samples can be balanced with TG data. A note has to be taken on disordered, X-ray amorphous or semi-graphite materials found in references, with regard to XRD analysis. All these materials could mean the same structural ordering, most probably a carbon phase in higher state than metaantracite, but containing only few nm thick graphite sequences, thus not producing detectable diffraction peaks. Such a material is detected by the amorphous hump method used in conjunction with the Rietveld refinement.
Based on our results, graphite is epigenetic, and due to the observed S content of graphite and V content observed in muscovite and in TiO2, it has organic material associated origin. The samples have metamorphic textures with the marks of shearing deformations, where graphite flakes and graphitic mixtures are mainly enriched. Thus, graphite was certainly formed in the shear zones during the regional metamorphism of the organic matter rich shales.
According to Árkai [21], Paleozoic formations of the Szendrő Mts went through on very low – lower middle grade of metamorphism: 300–450 °C and 250–300 MPa. Based on the observed mineral assemblages and crystallinity degree of sericite, illite and graphite, the grade of metamorphism is in the quartz-albite-muscovite-chlorite subfacies within the greenschist facies. According to our calculations from Raman spectroscopy results, the formation temperature, estimated by the formula of Beyssac et al. [18], ranges between 385 and 448 °C (± 30 °C). The average formation temperature, based on the average of 18 measurements, is around 410 °C (± 30 °C) [14]. This temperature value is in accordance with the above- mentioned values of metamorphism, this formula does not take into account any existing strain in the system. Deduced field of graphite crystallization from the thermoanalytical investigations in [17] is in the low pressure (< 50 MPa) medium temperature (450–500 °C) range.
Conclusions
Thermal analysis was a required method in extension of XRD and Raman spectroscopy to fully characterize the type, structural state and amount of carbon materials. On the other hands, for correct interpretation of DTA peaks and related TG values, Raman spectroscopy was useful in identifying the non-crystalline fraction of carbon.
All observations point to epigenetic graphite with probable organic material origin, supported by S content. It was certainly formed in low intensity shear zones (410–500 °C, < 50 MPa) during the regional metamorphism of the organic matter-rich shales. Its amount is 1.5–3 mass% in the samples calculated by Rietveld refinement, but considering the disordered ≈ X-ray amorphous or ≈ semi-graphite fraction as total graphitic material — as seen by DTA — then severe underestimation is observed compared to XRD-based crystalline graphite. However, considering amorphous fraction from XRD, very similar results are obtained by the two methods.
Altogether, application of DTA-TG is a necessary method to support and approve the compositional data on graphite bearing low metamorphic grade black schists.
References
Dill HG. The “chessboard” classification scheme of mineral deposits: Mineralogy and geology from aluminum to zirconium. Earth Sci Rev. 2010;100:1–420.
Reynolds WN. Physical properties of graphite. Elsevier, Amsterdam: Mater Sci Ser; 1968.
Mitchell CJ. Industrial Minerals Laboratory Manual: Flake Graphite. BGS Technical Report, Keyworth, Nottingham, WG/92/30; 1993.
Ji H, Chung Y, Pan X, Hua M, Shu C, Zhang L. Study of lithium-ion battery module’s external short circuit under different temperatures. J Therm Anal Calorim. 2020. https://doi.org/10.1007/s10973-020-09506-0.
Dhandayuthabani M, Jegadheeswaran S, Vijayan V, Godwin AA. Investigation of latent heat storage system using graphite micro-particle enhancement. J Therm Anal Calorim. 2020;139:2181–6.
Qian Y, Zhang Y, Sun J, Song C, Jing Y, Shao F, Jia Y, Tao Z, Wang X, Liu H. The effect of hydrophilic modification of expanded graphite on the thermophysical properties of magnesium chloride hexahydrate. J Therm Anal Calorim. 2020;140:1725–35.
Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on the 2017 list of Critical Raw Materials for the EU. European Commission. Brussels; COM(2017) 490 final.
U.S.G.S. Mineral Commodity Summaries 2020: Graphite (natural). U.S. Geological Survey, 2020. pp. 72–73. https://pubs.usgs.gov/periodicals/mcs2020/mcs2020-graphite.pdf Accessed 18 April 2020.
Kwiecińska B, Petersen HI. Graphite, semi-graphite, natural coke and natural char classification – ICCP system. Int J Coal Geol. 2004;57:99–116.
Pasteris JD, Wopenka B. Raman spectra of graphite as indicators of degree of metamorphism. Can Mineral. 1991;29:1–9.
Wopenka B, Pasteris JD. Structural characterization of kerogens to granulite-facies graphite: applicability of Raman microprobe spectroscopy. Am Mineral. 1993;78:533–57.
Fülöp J. Geology of Hungary, Paleozoic II. (In Hungarian). Budapest: Akadémiai Kiadó; 1994. pp. 9–118.
Majoros L, Kristály F, Szakáll S. Graphite in black schists from Dédestapolcsány (Uppony Mts.), Gadna and Szendrőlád (Szendrő Mts.) in NE-Hungary. Joint 5th Central-European Mineralogical Conference and 7th Mineral Sciences in the Carpathians Conference, Banská Štiavnica, Slovakia, 2018. Book of abstracts, p. 69.
Majoros L, Kristály F, Fintor K. Origin of graphite in black schists from Szendrő Mts (NE-Hungary), 1st International Student Conference on Geochemistry and Mineral Deposits. Prague, Czech Republic: Book of abstracts; 2019. pp. 33–4.
Hermesz M. (ed.) Geological and geophysical analysis of graphite bearing occurrences in the Szendrő Mts. (In Hungarian). TIT. Nógrád megyei Szervezete Környezetvédelmi és Műszaki Szakszolgálat Munkacsoportja. Unpublished report. National Geological and Geophysical Repository, Budapest; 1990.
Raincsákné Kosáry Zs. Devonian rocks of the Szendrő Mts. (In Hungarian). Geologica Hungarica, Series Geologica, 1978;18:1–113.
Gaweda A, Cebulak S. The origin of graphite in the crystalline basement of the Western Tatra Mts. (Western Carpathians, S-Poland). Bratislava: Geologica Carpathica, 1999;50:4:295–303.
Beyssac O, Goffé B, Chopin C, Rouzaud JN. Raman spectra of carbonaceous material in metasediments: a new geothermometer. J Metamorph Geol. 2002;20:859–71.
Gyalog L, Síkhegyi F. (series editor). Geological Map of Hungary, 1:100 000. Published by the Hungarian National Geological Institute, Budapest; 2005. https://map.mbfsz.gov.hu/fdt100/ Accessed 9 April 2020.
MBFSZ maps: Drills in Hungary. https://map.mbfsz.gov.hu/furas/ Accessed 9 April 2020.
Árkai P. Low-grade metamorphism of Paleozoic sedimentary formations of the Szendrő Mts. (NE-Hungary). Acta Geologica Academiae Scientiarum Hungaricae, 1977;21:53–80.
Downs RT, Hall-Wallace M. The American Mineralogist Crystal Structure Database. Am Miner. 2003;88:247–50.
Földvári M. (2011): Handbook of thermogravimetric system of minerals and its use in geological practice. Budapest: Occasional Papers of the Geological Institute of Hungary; 2011; vol. 213.
Funding
Open access funding provided by University of Miskolc. The research was carried out as part of the “More efficient exploitation and use of subsurface resources” project of the University of Miskolc, implemented in the framework of the Thematic Excellence Program funded by the Ministry of Innovation and Technology of Hungary. (Grant Contract reg. nr.: NKFIH-846–8/2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majoros, L., Fintor, K., Koós, T. et al. Metamorphic graphite from Szendrőlád (Szendrő Mts., NE-Hungary) detected by simultaneous DTA-TG. J Therm Anal Calorim 147, 3417–3425 (2022). https://doi.org/10.1007/s10973-021-10713-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-021-10713-6