
Abstract
The yttrium citrate was synthesized using freshly precipitated yttrium hydroxide and citric acid (or monosodium citrate) in the aqueous environment by the hydrothermal treatment at 80 °C or 100 °C. The properties of the samples were investigated using the CHN, XRD, TGA and static light scattering. Crystalline yttrium citrate dihydrate is formed by the reaction of citric acid with freshly precipitated yttrium hydroxide after 14 days of reaction at 80 or 100 °C. The thermogravimetric analysis of the crystalline sample of yttrium citrate dihydrate, thermal decomposition of the compound in the synthetic air occurs in four stages with the release of mainly water and carbon dioxide. Methane and propyne are formed in smaller amounts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Yttrium citrate is a precursor used for the production of various functional yttrium oxide coatings, doped with rare earth elements yttrium oxides, mixed oxides, ceramics [1,2,3,4,5]. In addition, yttrium citrate radiocolloids are used to treat the joint pain, e.g., rheumatoid arthritis [6,7,8]. Rheumatoid arthritis caused by inflammation of the synovium (thickening and swelling of the synovitis) invades cartilage and destroys the joint capsule and bone, so that it may be necessary to remove the synovial membrane, to restore the functionality of the joint. The alternative methods in relation to the surgical synovectomy are chemical synovectomy using thiotepa, nitrogen mustard, methotrexate, osmic acid or antibiotics [9] and synovectomy using radioactive isotopes deposited on the colloidal carriers (radiosynectectomy). Recently, good treatment results have been obtained by combining the method of surgical synovectomy and radiosynectomy [10, 11]. The method of radiosynectectomy involves injecting a radioactive colloid into a sick joint where radiation destroys the top layers of the membrane and reduces the number of blood vessels in it. The European Association of Nuclear Medicine indicates in Guideline for Radiosynovectomy the following radiocolloids: 90Y silicate or citrate, 186Re sulfide and 169Er citrate [12]. To avoid radioisotope leakage beyond the treatment area, radioisotopes are administered in the form of insoluble salts or are encapsulated [13]. Reducing the particle size is advantageous from the point of view of homogeneity irradiation; however, small particles promote the leakage and spread of the isotope. However, in the papers describing the application of radiocolloids used in radiosynovectomy, the sizes of radiocolloid particles differ significantly even for the same radioisotope [14,15,16]. In the literature, there is no agreement what size of particles is optimal in radiosynovectomy. According to EANM, 5–10-μm particles do not cause leakage [10] while van der Zant et al. consider 2–5 μm as the optimal size [3]. However, C. Cruz-Esteban believes that the ideal particle size is 100 nm [17]. In view of the above, it seems that one should use methods of making for monodisperse colloids for synovectomal colloids and characterize the dispersions by investigating the particle size distribution.
Isotope synovectomy (radiosynothesis) using colloids (citrate or silicate) labeled with 90Y is an effective method recommended for the local treatment of chronic inflammation of the knee joints [18]. The first treatment with the use of the 90Y isotope was made in 1968 to replace the 198Au colloids, which due to the emission of γ radiation of 411 keV energy induced undesired radiation [8]. Yttrium citrate is a common 90Y carrier in radiosynovectomy.
The methods of monodisperse colloids synthesis are based on the control of the nucleation process followed by the crystal growth process. Monodisperse particles are characterized by the same size, shape and composition. Their particle size distribution is characterized by a 10% relative standard deviation [19]. Numerous methods of obtaining monodisperse colloidal systems of various compounds or elements have been described in the literature. The most common methods that have been applied, among others, to metal oxides, meta-sulfides and metal selenides, can be divided into: precipitation from homogeneous solutions, phase transformations and aerosol reactions [20]. In the homogeneous precipitation methods to obtain homogeneous particles, forced hydrolysis, controlled release of anions or controlled release of cations was used. The structure and properties of yttrium citrates were studied initially for the separation of this element from other rare earth elements. The subsequent research concerned obtaining yttrium citrate for the purposes of synovectomy and in the synthesis of nanoscale yttrium oxides. In the literature reports, yttrium citrate was obtained by reacting citric acid with 90Y ions eluted from ionite (to separate from 90 Sr) [21]. Yttrium citrate was obtained by mixing 0.1 mol dm−3 of yttrium chloride with trisodium citrate, maintaining the pH in the range from 7 to 8. The sediment obtained in this method contained a variable number of water molecules from 1 to 3 [22]. Lanthanum citrate and an amount of lanthanide citrate were obtained by reacting the oxide with citric acid [22, 23].
This paper attempts to optimize the conditions for the synthesis of monodisperse yttrium citrate by transforming yttrium hydroxide to yttrium citrate in the reaction of freshly precipitated yttrium hydroxide with citric acid or its salts. To determine the preliminary synthesis conditions, the analysis of the solubility of yttrium hydroxide and yttrium citrate was carried out. These diagrams are based on the constant durability of yttrium complexes, yttrium citrates and the product of the solubility of yttrium hydroxide and yttrium citrate available in the literature.
In the literature, there are described/characterized several yttrium citrates compounds, two of which are complex compounds [24]:
where Cit−3 = C6H5O −3.7
The solubility product of the compound YCit*5H2O [22] is:
In the aqueous solutions between the reactions of dissolution of yttrium hydroxide and precipitation of yttrium citrate, there should be taken into account the following reactions: dissociation of citric acid [25], formation of hydroxides with yttrium, solubility product of yttrium hydroxide. Based on the literature data [23,24,26], the solubility of yttrium citrate and yttrium hydroxide as a function of pH was calculated; Fig. 1 illustrates these relationships.

A comparison of the solubility diagrams of yttrium hydroxide and yttrium citrate
As one can see, the minimum solubility of citrate falls in pH range from 4.3 to 9.2, while that of yttrium hydroxide at pH 10.1, and at the pH above 7.00 yttrium hydroxide is a more stable phase than yttrium hydroxide. In addition, it is considered that complexes of basic citrates may be formed at pH above 6.5 [22]. The dependencies shown in Fig. 1 indicate that the transformation of yttrium hydroxide into yttrium citrate is possible at pH below 7.0. It should be noted that the pH of the minimum solubility of yttrium citrate corresponds to the pH values of synovial fluid, which are in the range of 7 to 7.5 [27, 28], so under these conditions, yttrium citrate has the lowest solubility. After application, the pH value of synovial fluid will not change.
Experimental
Preparation of yttrium hydroxide
The first stage in this method of obtaining yttrium citrate was the synthesis of yttrium hydroxide as an intermediate product according to the procedure given by Brzyska [29]. 60 mL of a 0.1 M solution of yttrium chloride was added to a beaker to which 0.1 M NaOH solution was also added in small portions until the pH reaches approximately 10 (pH = 10.1) and a precipitate was formed. Next, the obtained precipitate was washed out with redistilled water on the filter in order to remove the sodium chloride (NaCl) formed during precipitation and to obtain a constant conductivity of the filtrate. The final conductivity was about 20 μS.
Description of the procedure for obtaining yttrium citrate
A 0.1 M citric acid solution was obtained by dissolution of citric acid (POCH S.A.) in redistiled water. The washed yttrium hydroxide was dispersed in a beaker in which 20 mL of 0.1 M citric acid solution was placed. The mixture was then sonicated for 3 min, the pH was measured, and the suspension was placed in the Teflon autoclave for a period of 14 days at the temperatures from 80 to 160 °C. The obtained sample of yttrium citrate was subjected to composition and structure analyses.
Methods
The elemental analysis of yttrium citrate samples for the C and H contents was conducted out using the PerkinElmer CHN analyzer. In addition to the quantitative analysis of the C and H contents, the thermogravimetric studies were carried out for the obtained yttrium citrate samples using the Netzsch STA 449 Jupiter F1 apparatus coupled with the Netzsch mass spectrometer and the Brucker FTIR spectrometer. The samples (20 mg) were heated at a rate of 10 °C min−1 in the range of 30–950 °C in the atmosphere of synthetic air (flow of 50 mL min−1). Alumina crucibles and samples carrier type S were used in the measurements. The MS measurement was performed in the scan mode of the range of m/z from 10 to 150, and the FTIR spectra were recorded in the spectral range of 650–4500 cm−1 with 16 scans per spectrum at a resolution of 4 cm−1. Data were collected and processed using the NETZSCH Proteus®. Diffraction measurements of the obtained samples were carried out by the PXRD (powder X-ray diffraction) method using the Panalytical Empyrean diffractometer. The radiation of the Cu lamp and SRM 676 external standard were used. The particle size distribution of the obtained preparations was determined by the method of static light scattering using the Malvern Mastersizer 2000.
Results and discussion
To obtain the crystalline citrate synthesis of yttrium citrate from yttrium hydroxide, the synthesis of yttrium citrate from freshly hydrated yttrium hydroxide and citric acid was carried out in a Teflon autoclave at the above-mentioned temperatures of 80–160 °C for 14 days. Only temperatures of 80 and 100 °C resulted in the crystalline deposit of yttrium citrate. The synthesis at higher than 100 °C led to amorphous samples. Table 1 presents the results of the C and H analyses for the samples obtained at 80 and 100 °C.
The synthesized samples contained slightly less C and slightly more H compared to YCit * H2O (24.35% C and 2.38%), but their composition was similar to YCit (22.93% C and 2.87%). The results of the TGA/FTIR and TGA/MS analyses of sample 1 are shown in Figs. 2 and 3.

TGA/FTIR spectra of sample No. 1

a TGA/MS spectra of sample No. 1. b TGA/MS spectra of sample No. 1
Analyzing the results presented in Figs. 2 and 3, one can see that the main products of thermal decomposition of sample No. 1 are: H2O (in the FTIR spectra shows the absorption band at 1508 cm−1 and in the MS spectrum the signal m/z = 18) and CO2 (in the spectrum of FIRT the band was observed at 2359 cm−1 and the MS spectrum signal at m/z = 44). In addition, weaker signals from CH4 can be found (in the FTIR spectrum the band was at 2997 cm−1 and in the MS spectrum signal was for m/z = 16).
The dependencies of DTG, H2O and CO2 FTIR spectra as well as MS spectra as a function of temperature show four areas presented in Figs. 4 and 5. As can be seen, there are four stages of the thermal decomposition process. This decomposition differs from the thermogravimetric analysis of yttrium citrate carried out by Petrova and Thodorovsky that indicates that the yttrium citrate complexes are stable up to 60–80 °C, and heating them to higher temperature leads to their decomposition [30]. The Petrova and Thodorovsky decomposition process proceeds in six stages partially overlapped. However, their sample of yttrium citrate prepared in the water medium or ethylene glycol was not stoichiometric. The first area in the temperature range of 150–217 °C is associated with a 4.2% loss of mass; FTIR spectra (Fig. 4) and MS spectra (Fig. 5) indicate the release of H2O and CO2 molecules that may be related to the physical adsorption of these molecules on the surface of yttrium citrate. In the temperature range 217–367 °C, the mass loss on the TG curve is 21.7%, it consists in the release of 2 molecules of crystalline water (11.47%), and in the FTIR spectrum there appears a band from CO2 at 2358 cm−1 and a weaker one for methane. This indicates a partial decomposition (oxidation of citrate ion). In this temperature range, Wiecinska observed release of H2O and CO2 as a result of thermal decomposition of citric acid [31]. The comparison of the peaks area in the MS spectrum (Fig. 5) shows that this range accounts for 8% of the total CO2, i.e., as a result of combustion/release and from 6 C atoms, statistically 0.5 C releases in the form of CO2. A similar comparison of the peak area m/z = 18 (for H2O) as a function of temperature indicates that 60% of water is released, i.e., statistically 2.7 molecules of water. It follows that some of the released water in this temperature range can come from the decomposition of citric acid to aconitic acid [32]. In the temperature range from 367 to 550 °C, the sample mass decreases to 39% of the initial mass. The analysis of the peak field in this range at 1508 cm−1 and the MS spectrum signal m/z = 18) as well as CO2 (in the FIRT spectrum the band was observed at 2359 cm−1 and the MS signal spectrum at m/z = 44) shows that 35% of H2O, i.e., about 1.6 particles and 82% C in the form of CO2, are released. The signals observed in this temperature range for m/z = 16 (CH4) and m/z = 40 (for C3H4 propene) are much weaker (Fig. 3) [32, 33]. In the TGA/FTIR and TGA/MS spectra of gases released during heating, the bands characteristic of itaconic anhydrite or citraconic anhydrite are not observed [32], although these compounds formed in the citrate decomposition process do not evaporate but oxidize in the air atmosphere. The sample heated to a temperature of 550 °C is characterized by a gray color, a crystal structure corresponding to yttrium oxide, but in the FTIR spectrum, the bands characteristic of HCO3 and OH groups are visible. Subsequent heating of the sample to 900 °C causes a mass decrease of approx. 5%, which is a result of oxidization of elemental carbon (as evidenced by the gray color of the sample heated to 550 °C) or decompositions of residual carbonates resulting from the disintegration/or oxidation of yttrium citrate. The MS spectrum of the characteristic peaks area for carbon dioxide shows 7.8%. A similar effect was observed by Petrova and Thodorovsky [30].

DTG/FTIR spectra of yttrium citrate after the 14-day synthesis using citric acid and freshly precipitated yttrium hydroxide

DTG/MS spectra of yttrium citrate after the 14-day synthesis using citric acid and freshly precipitated yttrium hydroxide
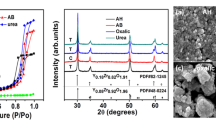
Results of diffraction measurements of crystalline sample of yttrium citrate.
Crystalline yttrium citrate was obtained as a result of a 14-day reaction of citric acid with freshly precipitated yttrium hydroxide in a Teflon autoclave at temperatures of 80 or 100 °C. An example of a diffraction pattern of a sample of yttrium citrate is shown in Fig. 6. The peaks of the resulting powder diffraction pattern were analyzed using the Dicvol program [34], and the obtained data are shown in Table 2 and the positions of the peaks are below the abscissa in Fig. 6, whereas the sets of cell parameters determined, M (20) and F (20) values [35, 36] for the studied samples are presented in Table 3. The analysis of Miller indices of diffraction reflections did not reveal any systematic absences, and hence, possible space groups are P2, Pm or P2/m. The average size of crystallites of sample No 1 using the peak maximum placement and full width at half maximum in the 2θ angle range from 9 to 23 deg and the Halder–Wagner method [37, 38] was 54.2 ± 3.7 nm. Using the Waxfit program, the determined crystallinity of the sample based on the analysis of the diffraction pattern in the 2θ range from 17 to 20 deg was 96.5%. Then, a sample of yttrium citrate heated to 550 °C was analyzed; the resulting X-ray powder diffraction pattern is shown in Fig. 7.

XRDP of yttrium citrate sample

XRDP of yttrium citrate sample heated up to 550 °C (brown line) and 850 °C (red line). (Color figure online)
The X-ray powder diffraction pattern of a sample of yttrium citrate heated to 550 °C indicates the presence of several peaks whose position corresponds to yttrium oxide the PDF card 1-88-1040. The analysis of the X-ray powder diffraction pattern by the Waxfit program showed that the degree of crystallinity of the sample is 67.3%, while the crystallite size estimated by the Halder–Wagner method is 4.0 ± 0.2 nm. Thus, heating the sample to a temperature of 550 results in the decomposition of yttrium citrate to yttrium oxide and the associated reduction in crystallites from 54 to 4 nm. Nevertheless, from the course of TG curves after fencing the sample above the temperature of 550 °C and the color of the sample after heating at 550 °C, it follows that in the samples after heating above 550 °C CO2 is released. The XRDP pattern of yttrium citrate heated to 850 °C shows more reflections and is sharper. Their position is consistent with the yttrium oxide PDF card 1-88-1040. The analysis of the X-ray powder diffraction pattern by the Waxfit program showed that the degree of crystallinity of the sample is 96.33% while the crystallite size estimated by the Halder–Wagner method is 15.6 ± 0.2 nm. To identify other compounds than yttrium oxide in the sample, the FTIR analysis of the sample heated to 550 °C was made.
As shown in Fig. 8 in the spectrum of the YCitr sample heated to 550 °C, the band at 3439 cm−1 is characteristic of the stretching vibrations of adsorbed water [39]. The bands at 1511, 1407 and 1075 cm−1 can be attributed to the stretching of the carbonate group and the band at 850 cm−1 to the stretching CO group, whereas the bands at 668 cm−1 and 463 cm−1 can be assigned to the stretching group YOH and the band 426 cm−1 associates with Y–O–Y bonds [40]. The FTIR spectrum indicates the presence of carbonate groups in the sample that can form basic yttrium carbonate. The XPS analysis was made out to determine the composition of the sample heated to 550 °C.

FTIR spectrum of the sample heated up to 550 °C
The survey scan depicted in Fig. 9a is composed of C 1s, O 1s and Y3d signals. The deconvolution of Y 3ds spectrum, (Fig. 9b) reveals four peaks at 160.4, 158.5, 157.6 and 156.9 eV. The peaks at 156.9 and 158.5 correspond to the spin–orbit splitting of Y 3d 5/2 and Y 3d 3/2 of Y in Y2O3 and two others at 157.6 and 160.4 eV for Y 3d5/2 and 3d3/2 of Y in yttrium carbonates [41, 42]. Figure 9c shows the C 1s band and the results of its deconvolution three components. The first peak located at 284.9 eV can be assigned to adventitious carbon, the second peak located at 286.2 eV (C–O–H, C–O–C), and the third peak located at 289.8 (O–C=O) [42]. These carbon oxygen species point out to the presence of carbonate compounds in the sample. The deconvolution of O1s band, depicted in Fig. 9d, results in three peaks. The first peak located at 529.4 eV is the nominal position of O1s in Y2O3, the second peak located at 531.9 eV can be attributed to the Y–OH group, and the third one located at 533.6 can be assigned to C–O–C=O [42]. The results of XPS analysis confirm the presence of carbonates and carbon in the YCit sample heated up to 550 °C.

a Full XPS spectrum of YCit heated up to 550 °C. b XPS spectrum of Y3d of YCit sample heated up to 550 °C. c XPS spectrum of C1s of YCit sample heated up to 550 °C. d XPS spectrum of O1s of YCit sample heated up to 550 °C
The particle size distribution of yttrium citrate.
Figure 10 depicts the particle size distributions of yttrium citrate sample obtained in the synthesis after 14 days. As can be seen, the sample obtained after the 14-day synthesis is characterized by a wide range of particle sizes from 0.5 to 800 μm. The comparison of the particle size for this sample with that of the crystallites indicates that the particles are a crystallites conglomerate.

Particle size distribution of crystalline (14—day synthesis) sample of yttrium citrate
Conclusions
The above presented study can be summarized as follows:
Crystalline yttrium citrate dihydrate is formed by the reaction of citric acid with freshly precipitated yttrium hydroxide after 14 days of reaction at 80 or 100 °C. The yttrium citrate is crystallized in a monoclinic system with the cell parameters a = 18.2286 Å b = 5.8226 Å c = 16.7186 Å and β = 93.09. The decomposition of the crystalline sample of yttrium citrate dihydrate in the synthetic air proceeds in four stages with the release of mainly water and carbon dioxide. Methane and propyne are formed in smaller amounts. In the first stage, physically adsorbed water and carbon dioxide are released from the surface of the compound; in the second the loss of water of crystallization and partial dehydration of citrate are observed. In the temperature range 350–550 °C, further dehydration and oxidation of citrate occur. Subsequent heating of the yttrium citrate to 550 °C releases carbon dioxide and semicrystalline yttrium oxide with the addition of amorphous yttrium carbonate, and elemental carbon is formed. Decomposition of the yttrium citrate by heating up to 850 °C leads to formation of crystal yttrium oxide of the size 15.6 ± 0.2 nm.
References
Chen J, Huang B, Huang C, Sun X. Preparation of nanoscaled yttrium oxide by citrate precipitation method. J Rare Earths. 2017;35:79–84. https://doi.org/10.1016/S1002-0721(16)60164-3.
Zhong S, Wang S, Liu Q, Wang Y, Wang Sh, Chen J, Xu R, Luo L. Y2O3:Eu3+ microstructures: Hydrothermal synthesis and photoluminescence properties. Mater Res Bull. 2009;44:2201–5. https://doi.org/10.1016/j.materresbull.2009.08.009.
Mangalaraja RV, Mouzonb J, Hedstromb P, Kerob I, Ramama KVS, Camurria CP, Odén M. Combustion synthesis of Y2O3 and Yb–Y2O3 part I. Nanopowders and their characterization. J Mater Process Technol. 2008;208:415–22. https://doi.org/10.1016/j.jmatprotec.2008.01.023.
Kruk A, Polnar J. Investigation on the physicochemical properties of La-doped Er0.05Y1.95O3 nanopowders. J Therm Anal Calorim. 2020;139:765–73. https://doi.org/10.1007/s10973-019-08499-9.
Moskal G, Mikuskiewicz M, Jasik A. Thermal diffusivity measurement of ceramic materials used in spraying of TBC systems. J Therm Anal Calorim. 2019;138:4261–9. https://doi.org/10.1007/s10973-019-08589-8.
Heuft-Dorenbosch LLJ, de Vet HCW, van der Linden S. Yttrium radiosynoviorthesis in the treatment of knee arthritis in rheumatoid arthritis: a systematic review. Ann Rheum Dis. 2000;59:583–6.
Miszczyk M, Jochymek B, Miszczyk L, Matysiakiewicz J, Spindel J, Jabłońska I, Mrożek T, Chrobok A, Tomasik P, Zakrzewski T, Tukiendorf A. The results of 394 consecutive cases of knee joint radiation synovectomy (radiosynoviorthesis) using 90Y. Ann Nucl Med. 2020;34:94–101. https://doi.org/10.1007/s12149-019-01418-w.
Mödder G. Radiosynoviorthesis (radiation synovectomy) in clinical nuclear medicine. In: Ahmadzadehfar H et al. editors. Clinical nuclear medicine. Springer Nature Switzerland AG 2020. p 1015–1024
van der Zant FM, Jahangier ZN, Gommans GGM, Moolenburgh JD, Jacobs JWG. Radiation synovectomy of the upper extremity joints: does leakage from the joint to non-target organs impair its therapeutic effect? Appl Radiat Isot. 2007;65:649–55. https://doi.org/10.1016/j.apradiso.2007.01.010.
Karaman A, Guney F, Dogar IH, Kafadar O, Bilal M, Oner Kula M. Comparison of arthroscopic, radioactive and combined synovectomy in the treatment of chronic non-specific knee synovitis. Med Princ Pract. 2014;23:551–5. https://doi.org/10.1159/000365635.
Nassar WAM, Bassiony AA, Elghazaly HA. Treatment of diffuse pigmented villonodular synovitis of the knee with combined surgical and radiosynovectomy. HSSJ. 2009;5:19–23. https://doi.org/10.1007/s11420-008-9104-5.
Clunie G, Fischerem M. EANM procedure guidelines for radiosynovectomy. Eur J Nucl Med. 2003;30:BP12–BP16.
Hafelia UO, Robertsa WK, Pauera GJ, Kraeftb SK, Macklisa RM. Stability of biodegradable radioactive rhenium (Re-186 and Re-188) microspheres after neutron-activation. Appl Radiat Isot. 2001;54:869–79. https://doi.org/10.1016/s0969-8043(00)00313-4.
Siegel ME, Siegel HJ, Luck JV Jr. Radiosynovectomy’s clinical applications and cost effectiveness: a review. Semin Nucl Med. 1997;27(4):364–71. https://doi.org/10.1016/S0001-2998(97)80009-8.
Karavida N, Notopoulos A. Radiation Synovectomy: an effective alternative treatment for inflamed small joints. Hippokratia. 2010;14(1):22–7.
Silva M, Luck JV Jr, Llinás A. Chronic hemophilic synovitis: the role of radiosynovectomy. Treat Hemoph. 2004;33:1–8.
Cruz-Esteban C, Wilke WS. Non-surgical synovectomy. Baillieres Clin Rheumatol. 1995;9(4):781–801 (ISBN 0-7020-2076-1).
Eur J Nucl Med.2003;30: BP12–BP16.
Sugimoto T. Mechanisms of nucleation and growth of monodispersed particles. In: Hubard, editor. Encyclopedia of surface and colloid science. New York: Marcel Dekker inc.; 2002. p. 3208–3218.
Matijevic E. Monodispersed colloids: art and science. Langmuir. 1986;2:212–20. https://doi.org/10.1021/la00067a002.
Bowen BM, Darracott J, Garnett ES, Tomlinson RH. Yttrium-90 citrate colloid for radioisotope synovectomy. Am J Hosp Pharm. 1975;32:1027–30 PMID:1190215.
Gmelin Handbook of Inorganic Chemitry—8th edn. Berlin: Springer; 1984. p 344–358.
Baggio R, Perec M. Isolation and characterization of a polymeric citrate. Inorg Chem. 2004;43:6965–8.
Zhou RS, Song JF, Yang QF, Xu XY, Xu JQ, Wang TG. Syntheses, structures and magnetic properties of a series of 2D and 3D lanthanide complexes constructed by citric ligand. J Mol Struct. 2008;877:115–22. https://doi.org/10.1016/j.molstruc.2007.07.027.
Lide DR, editor. CRC handbook of chemistry and physics. 88th ed. Boca Raton: CRC Press/Taylor &Francis Group; 2007.
Spahiu K, Bruno J. A selected thermodynamic database for REE to be used in HLNW performance assessment exercises. SKB Technical Report 95-35., Stockholm 1995.
Jebens EH, Monk-Jones ME. On the viscosity and pH of synovial fluid and the pH of blood. J Bone Joint Surg Br. 1959;41:388–400 PMID:13641329.
Cummings NA, Nordby GL. Measurement of synovial fluid pH in normal and arthritic knees. Arthritis Rheum. 1966;9:47–56. https://doi.org/10.1002/art.1780090106.
Brzyska W. Lantanowce i aktynowce. Warszawa: WNT; 1987. p. 75.
Petrova N, Todorovsky D. Thermal decomposition of zirconium–yttrium citric complexes prepared in ethylene glycol and water media. Mater Res Bull. 2006;41:576–89. https://doi.org/10.1016/j.materresbull.2005.09.007.
Wiecinska P. Thermal degradation of organic additives used in colloidal shaping of ceramics investigated by the coupled DTA/TG/MS analysis. J Therm Anal Calorim. 2016;123:1419–30. https://doi.org/10.1007/s10973-015-5075-1.
Wyrzykowski D, Hebanowska E, Nowak-Wiczk G, Makowski M, Chmurzynski L. Thermal behaviour of citric acid and isomeric aconitic acids. J Therm Anal Calorim. 2011;104:731–5. https://doi.org/10.1007/s10973-010-1015-2.
Daly NJ, Ziolkowski F. Thermal decomposition of citraconic anhydride. Aust J Chem. 1971;24(4):771–5.
Boultif D, Louer D. Powder pattern indexing with the dichotomy method. J Appl Cryst. 2004;37:724–31. https://doi.org/10.1107/S002188980401487.
Wolff PM. A simplified criterion for the reliability of a powder pattern indexing. J Appl Cryst. 1968;1:108–13.
Smith GS, Snyder RL. FN: A criterion for rating powder diffraction patterns and evaluating the reliability of powder-pattern indexing. J Appl Cryst. 1979;12:60–5. https://doi.org/10.1107/S002188987901178X.
Oleszak D, Olszyna A. Crystallite size and lattice strain determination of NiAl–Al2O3 nanocomposite from X-ray diffraction line broadening. Kompozyty (Composites). 2004;4:11–8.
Halder NC, Wagner CNJ. Separation of particle size and lattice strain in integral breadth measurements. Acta Crystallogr. 1966;20(2):312–31. https://doi.org/10.1107/S0365110X66000628.
Xiaoxu W, Yemin H, Xianghai M, Ying L, Mingyuan Z, Hongming J. Synthesis of Y2O3 phosphor by a hydrolysis and oxidation method. J Rare Earths. 2015;33:706–11. https://doi.org/10.1016/S1002-0721(14)60474-9.
Aghazadeh M, Barmi AM, Mohammad H. Cathodic electrodeposition of Y(OH)3 and Y2O3 nanostructures from chloride bath. Part II: effect of the bath temperature on the crystal structure, composition and morphology. Ceram Int. 2013;39(2):1045–55. https://doi.org/10.1016/j.ceramint.2012.07.026.
Gougousi T, Chen Z. Deposition of yttrium oxide thin films in supercritical carbon dioxide. Thin Solid Films. 2008;516:6197–204. https://doi.org/10.1016/j.tsf.2007.11.104.
Lin L, Starostin SA, Li S, Khan SA, Hessel V. Synthesis of yttrium oxide nanoparticles via a facile microplasma-assisted process. Chem Eng Sci. 2018;178:157–66. https://doi.org/10.1016/j.ces.2017.12.041.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janusz, W., Skwarek, E., Sternik, D. et al. Synthesis and thermogravimetric analysis of monoclinic yttrium citrate dihydrate. J Therm Anal Calorim 146, 1027–1038 (2021). https://doi.org/10.1007/s10973-020-10107-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-10107-0