
Abstract
Detailed vibrational (IR, Raman, far-IR) and thermal (TGA, TG–MS, DSC) analysis has been performed on di[κ1O,κ2O-carbonatotetraamminecobalt(III)] sulfate trihydrate, ([Co(NH3)4CO3]2SO4·3H2O (1). Its isothermic heating at 100 °C leads to formation of [Co(NH3)4CO3]2SO4 (compound 2). UV and IR studies showed that the distorted octahedral arrangement around cis-O2CoN4 core in compound 1 does not change during dehydration, which explains the reversible water loss and ability of compound 2 to rehydrate into compound 1. Compound 2 decomposes at ~ 240 °C in inert atmosphere giving final decomposition products, which are two modifications of nanosized metallic cobalt (hcp-15 nm, fcc-250 nm) and CoO (55 nm). The redox reaction results in N2 as an ammonia oxidation product. The decomposition intermediate is a cobalt(II) compound, Co2O1,14+δ(SO4)0.86 (δ = the oxygen surplus due to the presence of 2.8% of Co(III) ion). The same reaction in air atmosphere resulted in Co2O1.25+δ(SO4)0.75 (δ = the oxygen surplus due to the presence of 5.3% of Co(III) ion (compound 3a). Compound 3a is oxidized in air at 793 °C into Co3O4. The compound 3a exhibits catalytic activity in photodegradation in Congo red. The photodegradation process follows pseudo-first-order kinetic (kapp = 1.0 and 7.0. at pH = 3.4 and 5.25, respectively).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To continue our studies on the solid-phase quasi-intramolecular redox reactions between complex cations having redox-active ligands or cations [1,2,3,4,5,6,7,8], our next target was the preparation of [Co(NH3)4CO3](XO4)n (X = oxidizing tetrahedral anion) type compounds as precursors for the preparation of various Co oxide catalysts [9, 10]. The [carbonatotetraamminecobalt(III)] sulfate trihydrate, ([Co(NH3)4CO3]2SO4·3H2O (1) has been known for a long time [11] and used as a water-soluble (s ≥ 60.61 g L−1 [12]) ionic sulfate compound as a precursor for the preparation of other type Co-complexes [13], or as a reagent in the detection of proteins [14] and for feeding of fungi [15] as well. Some of the early investigators described the crystals of compound 1 with very different habit and structure as rhombohedral [16], orthorhombic [17] or monoclinic [18, 19] material. The single-crystal study confirmed the existence of the monoclinic form of compound 1 [20] at room temperature.
Amigo et al. [21] studied the decomposition of compound 1 in air until 800 °C, while Onodera et al. [22] combined TG, DSC and GC studies performed in inert atmosphere till 400 °C. The results given in these papers are very different, which indicates that the nature of the atmosphere has a crucial influence on the decomposition pathway. The diversity of crystal habits and the contrary information about the thermal behavior of compound 1 initiated us to study the existence of polymorphism and thermal behavior of it, including the identification of thermal decomposition products and intermediates as well.
In this work, the thermal decomposition characteristics of compound 1 in air and inert atmosphere with DSC and TG–MS were studied in detail. The nature of decomposition products and the influence of the atmosphere on the decomposition processes were also studied. The compound 1, its dehydrated form ([Co(NH3)4CO3]2SO4, compound 2) and the earlier unknown thermal decomposition intermediate formed in the decomposition of compound 1 in air (basic cobalt(II) sulfate, Co2O1.25+δ(SO4)0.75, δ = the oxygen surplus due to presence of 5.3% of Co(III) ion in the compound), compound 3a) were characterized by spectroscopic methods. The photocatalytic activity of compound 3a on the degradation of toxic dyes is presented.
Experimental
Deuton-X Ltd., Hungary, supplied chemical-grade cobalt carbonate, ammonium hydroxide (25%) and ammonium carbonate, hydrogen peroxide (30%) and analytical grade of perchloric acid, sodium hydroxide, ethanol, barium hydroxide, barium nitrate, hexachloroplatinic acid hexahydrate, basic cobalt(II) carbonate and sulfuric acid.
Preparation
To prepare compound 1, a modified method described by Jorgensen [25] was followed by dissolving 20 g of cobalt carbonate in dilute H2SO4 (10%), reaching a total volume of 100 mL. Then, the clear solution was poured into a solution containing 100 g of (NH4)2CO3, and 500 mL of distilled water and 250 mL of cc. ammonia and oxidation were performed during 3–4 h through a constant oxygen stream system. After the oxidation step was finished, a blood-red solution was expected to contain several small pieces of (NH4)2CO3. Evaporation proceeded on a steam bath until it reaches the volume of 300 mL, and then, the solution was filtered out. The solution was evaporated again to turn into a more concentrated form (200 mL) and chilled out, whereupon [Co(NH3)4CO3]2SO4·3H2O crystallizes as purplish-pink prisms. The mother liquor was decanted, and the precipitates were filtered off (washed with a saturated solution containing a small portion of the precipitate). Further evaporation of the mother liquor resulted in more salt formation. It is essential to add some (NH4)2CO3 during the evaporation step.
The anhydrous salt (compound 2) was prepared with isothermal heating of compound 1 at 120 °C for 2 h in air. Similarly, the compounds 3a and 3b were prepared at 300 °C in air or N2, respectively, for 2 h in both cases.
Elemental analysis
The sulfate ion content was determined gravimetrically. Dissolution of ca. 1 g precisely weighted sample in 20 mL water, precipitating the barium sulfate in a usual way with 2 equivalent of barium nitrate dissolved in water. The processing was done in the usual way [30]. To determine ammonia and carbonate content in the compound 1, the solid compound 1 was put into a three-necked flask equipped with a thermometer and dropping funnel, and 20 mL of 10% of NaOH solution was added dropwise under heating to release all the ammonia from the solution. The evolved ammonia was absorbed and precipitated as ammonium hexachloroplatinate, filtered off and heated at 500 °C to transform that into metallic platinum. The solution left back contains cobalt(III) hydroxide precipitate and sodium carbonate was acidified, and the liberated CO2 was precipitated with Ba(OH)2 as BaCO3 and measured according to the usual way [30]. The cobalt content of the solution was measured as Co3O4, oxidizing the cobalt content of the sample (in case of compound 3 after dissolution in perchloric acid) with sodium peroxodisulfate then precipitating the Co(OH)3 and heating that at 950 °C for 2 h. The cobalt(III) content determination has been done according to the known method with iodometry [31]. The ammonium sulfate content of the samples was decomposed by boiling them with 10% aq. NaOH solution for 30 min under N2 atmosphere.
Instrumental methods
X-ray powder diffraction measurements were taken using a Philips PW-1050 Bragg–Brentano parafocusing goniometer. It was equipped with a Cu tube operated at 40 kV and 35 mA tube power, a secondary beam graphite monochromator and a proportional counter. Scans were recorded in step mode with 0.04° step size for 1 s between 25° and 65° 2θ. Evaluation of the diffraction patterns had been obtained by full profile fitting techniques. The in situ, high-temperature XRD measurements were taken in a nitrogen atmosphere in a high-temperature HTK-1200 Anton–Paar chamber with temperature programming, at 450, 550 and 800 °C, respectively.
FTIR measurements were recorded on a Jasco FT/IR-4600 system, equipped with a Jasco ATR Pro One single reflection diamond ATR accessory (incident angle 45°), and a DLATGS detector in the 4000–400 cm−1 region. A resolution of 4 cm−1 and co-addition of 64 individual spectra was applied. Before the evaluation, an ATR correction (Jasco Spectra Manager version 2, Spectra analysis module version 2.15.11) was performed on the raw spectra. Far-IR spectra were recorded on a BioRad-Digilab FTS-60A spectrometer with 6.25 Mylar beamsplitter equipped with Pike GladiATR accessory with diamond ATR crystal for the 700–40 cm−1 range in Nujol mull.
The Raman measurements were taken using Horiba Jobin–Yvon LabRAM-type microspectrometer with external 532 nm Nd-YAG (~ 40 mW) and 785 diode laser sources (~ 50 mW) and Olympus BX-40 optical microscope. Optional optical density filters (D = 0.3 and 0.6) were applied to decrease laser light intensity avoiding the thermal degradation of samples. The laser beam was focused by an objective of 20 × (NA = 0.4). The confocal hole of 1000 µm and 950 (for 785 nm light) and 1800 groove mm−1 (for 532 nm light) grating monochromators were used in a confocal system and for light dispersion. In the case of 532 nm excitation, the spectral range of 100–4000 cm−1 was detected as the relevant range with 3 cm−1 resolution collecting the spectrum for 120 s per point. In the case of 785 nm excitation, the measured spectral range was between 100 and 2400 cm−1 with 5 cm−1 resolution and the exposure time of the sample was 30 s.
Thermal data in air and nitrogen were collected using TA Instruments SDT Q600 thermal analyzer coupled to Hiden Analytical HPR-20/QIC mass spectrometer. The decomposition was followed from room temperature to 500 °C at 10 K min−1 heating rate in nitrogen and air as carrier gas (flow rate = 50 cm3 min−1). Sample holder/reference: alumina crucible/empty alumina crucible. Sample mass 1–2 mg. The coupled TG–MS measurements were taken by heating rate 5 K min‒1 in argon and air. Selected ions between m/z = 1–97 were monitored in Multiple Ion Detection Mode (MID).
Simultaneous thermogravimetric, differential scanning calorimetric and mass spectrometric evolved gas analysis (TG–DSC–MS) measurements under inert conditions were taken on a Setaram LabsysEvo thermal analyzer, in high-purity helium (6.0) atmosphere, with a flow rate of 90 mL min−1. The measurements were recorded in the 25–500 °C temperature range, with a heating rate, of 20 °C min−1, and samples were weighed into 100-µL alumina crucibles. The obtained data were baseline corrected and further evaluated by the thermoanalyzer’s processing software (Calisto Processing, ver. 2.01). Parallel to the TG–DSC measurement, the analysis of the evolved gases/decomposition products was carried out on a Pfeiffer Vacuum OmniStar™ quadrupole mass spectrometer coupled to the above-described TGA. The gas splitters and transfer lines to the spectrometer were thermostated to 220 °C. The measurements were taken in SEM Bargraph Cycles acquisition mode, in which the total ion current (TIC), the analog bar graph spectra (for structure determination) and the separate ion current of each scanned individual mass (96 masses) were recorded. The scanned mass interval was 5–100 amu, with a scan speed of 20 ms amu−1, and the spectrometer was operated in electron impact mode.
Nitrogen physisorption measurements using Quantachrome Autosorb 1C static volumetric apparatus at − 196 °C. Samples of cc. 0.1 g were outgassed under vacuum before measures at 110 °C for 24 h. The specific surface area was calculated by the BET method.
The morphologies of the product were studied by ZEISS EVO 40XVP scanning electron microscope (SEM) operating at 20 kV. The SEM measurements were taken with different magnifications.
Photocatalysis
To evaluate the photocatalytic activity of the compounds 3a and Co3O4 prepared at 300 and 750 °C, respectively, under an oxidizing atmosphere, 1.0 mg of each Co-containing material was put into 3 mL of an aqueous solution of Methyl Orange (4 × 10−5 M) and Congo Red (2 × 10−5 M) dyes by using quartz cuvettes. The samples were kept in the dark overnight for the adsorption equilibrium. After that, they were submitted to a UV radiation source by Osram 18-W blacklight lamps (λ = maximum intensity at 375 nm). The cuvettes were placed 5 cm from each lamp, and the absorbance was measured every 30 min during 4 h by a Jasco V-550 UV–Vis spectroscope. The relative absorbance values of the most intensive peaks for Methyl Orange (464 nm) at pH 5.60 and Congo Red (497 nm) at pH 5.73 were considered to evaluate the catalysts’ activity in the degradation of dyes.
Preparation and properties of compounds 1, 2 and 3
The compound 1 was prepared first by Vortman [11, 23] in the reaction of basic cobalt carbonate with ammonia and ammonium carbonate on heating in air for 2 days, reacting the formed mixture with cold dilute sulfuric acid. The elemental analysis showed that compound 1 crystallizes with three molecules of water per formula unit confirmed by Benrath and Kohlberg [24] by vapor pressure measurements. Jörgensen [25] improved the method, adding cobaltous carbonate dissolved in diluted sulfuric acid to a solution of aqueous NH3-ammonium carbonate mixture, passing a current of air through the liquid for 2–3 h. Uspensky and Chibisov [26] obtained compound 1 by passing carbon dioxide gas into an aqueous solution of diaquotetraamminecobalt sulfate, in the presence of calcium carbonate. The purity of compound 1 depends heavily on the experimental conditions because the isomeric (Co(NH3)4SO4]CO3·2H2O [23, 27], and diaquotetraamminecobalt(III) sulfate/carbonate compounds [28, 29] or penta- and hexaamminecobalt(III) compounds with carbonate and sulfate counter ions [25] are also formed under similar experimental conditions. In our experimental methodology, the Jörgensen’s method [25] was modified by bubbling oxygen gas instead of air. The ruby-red crystals showed strong pleochroic behavior—the color parallel to (100) is purple-red while that parallel to (010) is brownish-red with a purple tinge, and that parallel to (001) is brownish-red [18]. The low-temperature DSC results under helium showed that the compounds 1 had not been any polymorphs between − 150 °C and its thermal decomposition point.
The composition and purity of compound 1 were checked by elemental analysis, XRD and IR spectroscopy to disclose the presence of others, e.g., sulfate-coordinated isomers. The ammonia complexes of cobalt(III) ion are stable enough toward hydrolysis; thus, the sulfate content could be measured directly as BaSO4 [23, 30]. The solution of compound 1 was decomposed with NaOH, and the liberated ammonia was precipitated as ammonium hexachloroplatinate [30]. The CO2 evolved on acidifying of mother liquor was measured as BaCO3 [30]. To determine the cobalt(III) content, the residual mother liquor was treated with sodium peroxidisulfate and NaOH, and the formed brown precipitate was measured as Co3O4 after heating at 950 °C for 2 h [30]. Compound 2 was analyzed in the same way, while compound 3 was dissolved in perchloric acid, and the cobalt(III) content was measured by iodometry [31].
Spectroscopic characterization (IR, far-IR, UV, Raman) of compound 1
The IR band assignations of the complex cation and the sulfate anion in complex 1 have already been published by Siebert [32] and Goldschmidt et al. [33, 34]. However, there have been no Raman measurements and factor group analysis on compound 1. The ammonia ligands coordinated to the cobalt(III)-ion forms hydrogen bonds with the sulfate and carbonate ions, determined from the values of the refractive indices [35], spin–lattice relaxation times/ammonia ligand reorientation energies [36] and single-crystal studies [20] as well. It can cause symmetry lowering, and thus, the number and types of vibrational modes for the cationic ([Co(NH3)4CO3]+), anionic (SO 2−4 ) and crystallization water (H2O) parts of the compound 1 were predicted by the factor group method. For the factor group analysis, the exact space group (P21/c) was taken from [20].
Vibrational modes of the cation
Octahedral skeleton
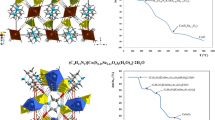
The cationic part of compound 1 has octahedral cis-O2CoN4 type coordination structure (distortion of CO 23 -ion symmetry from D3h to C2v), the carbonato group fills two coordination sites, and the other four coordination sites are occupied with ammonia molecules. There are two different crystallographic types of central cobalt(III) ions. Thus, the total number of modes for the Co3+ cations is doubled and equals 2 × (4 × 3) = 24 translational degrees of freedom (Fig. 1). All modes are Raman and IR active (Fig. 1).

Factor group analysis results of central Co3+ cation in compound 1
There are three types of stretching and bending modes for a cis-O2CoN4 type octahedral cation (NCoN, NCoO and OCoO), which have considerable coupling [33]. The measured far-IR and Raman spectroscopic data and the calculated frequency values for an isolated [Co(NH3)4CO3]+ ion (fCoN = 1.6 and fCoO = 1.25 [33]) are given in Table 1.
These assignments are reasonable as tentative assignments, based on the common assumption that M–N stretchings are higher than the corresponding M–O stretchings. Logically, for the stretchings of MN2, MO2 and MON, one would expect the following order of wavenumbers νs(NMN) > νs(NMO) > νs(OMO), and also νas > νs for all of the above species. The CoO2 moiety (C2v) in [Co(NH3)4CO3]+ complexes have higher wavenumbers for antisymmetric Co–O stretching frequency values than symmetric Co–O one [33, 38], thus among the two observed one, the band observed at 396 cm−1 in IR and 404 cm−1 in Raman spectra were assigned as the antisymmetric while the lower frequency bands at 335 and 318 cm−1 in the IR and Raman spectra, respectively, were assigned as the bands of the symmetric Co–O mode. Each Co–N band belongs to CoN2, and CoNO moieties cannot be distinguished unambiguously, partly due to symmetry lowering of the regular octahedron, which contains cis-coordinated carbonato-group, and partly due to the differences in Co–N and Co–O bond distances and consequently the mixed character of these bands.
Vibrational modes of the coordinated ammonia in compound 1
Due to the two crystallographic different [Co(NH3)4CO3]+ cation of the structure, eight crystallographic different ammonia molecules could be distinguished. There are four internal vibrational modes of NH3 molecules under C3v symmetry, ν1 is the symmetric stretching vibration (νs)(A1); ν2 is the symmetric bending (δs(A1); ν3 is the antisymmetric stretching vibration (νas(E); ν4 is the antisymmetric bending [δas(E) (Fig. 2)]. The ν3, ν4 and Txy and Rxy external mode are doubly degenerated modes under C3v while Tz and Rz are non-degenerated ones) (ESI Fig. 1).

Factor group analysis of internal modes of ammonia molecules (eight types) in compound 1
The total number of factor group modes due to the internal and external vibrations is the same, 8 × 24 for eight crystallographic types of NH3 in each case that is equal to 192 internal and 192 external (96 due to hindered translation and 96 due to hindered rotations) vibrational degrees of freedom. The degeneracies are lifted, and all the modes are IR and Raman active. The rocking mode of NH3 (ν5(E), ρ(NH3)) belongs to the Co–NH3 fragment, and the νs(CoN)(A1) mode of this fragment is discussed among translational modes of Co3+ ion (Table 2).
Using a laser with a 785 nm excitation beam did not give NH band intensities enough to evaluate, but at 532 nm excitation, we could get a Raman spectrum in the range of NH bond modes. The broad bands of symmetric and antisymmetric NH modes are strongly overlapped (Table 2). The Raman spectra recorded at 532 nm excitation gave a combined band containing symmetric HNH bending and antisymmetric CO stretching modes as a complex band system at 1293 cm−1. In the Raman spectra recorded at 785 nm, only the Co–O antisymmetric stretching mode component of the band system appears and is centered at 1273 cm−1. It is confirmed by the shifting of HNH mode of the band system centered at ~ 1300 cm−1 in the IR spectrum (ESI Fig. 2) when a component of the band after deuteration of the sample is left back at 1265 cm−1 (Co–O mode), while the higher wavenumber component is shifted to 1020 and 1004 cm−1 (symmetric ND2 bending mode).
The C=O§ and δas(HNHN) mixed band centered at 1645 cm−1 decomposes into components on deuteration, and the C=O§ component is located at ~ 1628 cm−1 in the perdeuterated 1, while the ND2 symmetric bending mode appears at 1162 cm−1. It means the δas(NH2) component is the higher wavenumber component of the mixed band. The C=O§ component is not sensitive on deuteration, and the deuteration caused only 1 cm−1 shift on the C=O§ in the band positions of the similar carbonatotetraamminecobalt(III) perchlorate complex [38].
Vibrational modes of the carbonate ion in compound 1
The free carbonate ion is planar (D3h) with four internal normal modes of vibration, namely ν1(A1′), (νs(C–O)), symmetric stretching), ν2(A2″) (π(CO3), symmetric bending), ν3(E′), (νas(CO)), doubly degenerate antisymmetric stretching) and ν4(E′) (δas(OCO)), doubly degenerate antisymmetric bending). The ν1, ν3 and ν4 are IR, while the ν1, ν3 and ν4 are Raman active. On coordination, either monodentate or bidentate(chelate) form, the symmetry of carbonate ion is lowered to C2v, and the selection rules are changed (Fig. 3). External modes of CO32− anions under D3h symmetry (T’s are hindered translations; R’s are hindered rotations (librations); modes of any of the E-symmetries are doubly degenerate. The results of factor group analysis for the two different types of carbonate ion can be seen on (ESI Fig. 1).

Factor group analysis of carbonate anions (two types) internal and external vibrations
The total number of factor group modes due to the internal (24) and external vibrations (24) of a carbonate anion is 48 (24 + 24), but because of two crystallographic types of carbonate anions that equals with 2 × 48 vibrational degrees of freedom (2 × 24 = 48 internal modes and 24 hindered translations and 24 hindered rotations) (Fig. 3, ESI Fig. 1). The degeneracies are lifted, and all internal and external vibrations are both IR and Raman active. The bands belong to the coordinated carbonate ion modes in compound 1, and their assignations are listed in Table 3. The stretching modes of non-coordinated oxygen (ν1) and coordinated ones (ν2(A1) and ν4(E), symmetric and antisymmetric one, respectively) can easily be distinguished. There are two kinds of in-plane OCO deformation mode, one with (ν5(B1) and one without (ν3(A1) involving of non-bound oxygen. The wavenumber of the highest stretching wavenumber of carbonate ion (1610 cm−1) belongs to the non-coordinated C=O bond, and the difference between this one and the higher component of the antisymmetric C–O (coordinated) mode (Δν = ~330 cm−1) unambiguously show the bidentate coordination mode of carbonate ion in compound 1.
The spectroscopic results disclose the monodentate carbonate ion coordination (ν(C=O§) should be found between 1450 and 1500 cm−1 and Δν value should not exceed 150 cm−1) and the outer sphere non-coordinated mode (D3h) carbonate ion which would be present in the sulfate-coordinated tetraamminesulfatocobalt(II) carbonate isomer (compound 6) [39].
Vibrational modes of the sulfate anion
The tetrahedral (Td) sulfate anion has four internal vibrational modes, namely the symmetric and antisymmetric S–O stretchings (ν1(A1) and ν3(F2)) and the symmetric and antisymmetric SO bending modes (ν2(E) and ν4(F2)). The symmetric bending mode is doubly, while both antisymmetric modes and the hindered rotations and translations of the sulfate anions are triply degenerate under Td symmetry. The total number of factor group modes due to the internal SO 2−4 vibrations equals 36 vibrational degrees of freedom (4 × 9).
The total number of factor group modes due to the external SO 2−4 vibrations (hindered translations and hindered rotations) equals 24 degrees of freedom (12, for the hindered translations and 12 for the hindered rotations of the sulfate anions). The factor group analysis results for the internal and external mode are summarized in Fig. 4 and ESI Fig. 1, respectively. All four internal and the external modes are IR and Raman active in compound 1, and all degeneracies are lifted (Table 4).

Internal modes of sulfate ion in compound 1
The band’s singlet nature at 971 cm−1 (IR) with low intensity compared with the intensive Raman bands confirms it as the symmetric S–O stretching mode because of the IR and Raman intensities of νs and νas bands appear in opposite relations in IR and Raman spectra. The symmetric one is weak (or forbidden in the IR) and quite intensive in the Raman spectra, while νas is very intense in IR but very weak in the Raman spectra of tetrahedral ions [1,2,3,4,5, 40]. The intensive band system centered at 1105 cm−1 (IR) with two shoulders (triplet nature) belongs to the antisymmetric S–O stretching mode, and according to this, a weak intensity band system could be observed (~ 1022 and ~ 1101 cm−1) in the Raman spectrum. The double degenerate symmetric OSO bending mode of sulfate ion can be assigned around ~ 483 cm−1 (IR) and 473 cm−1 (Raman). This band is also much more intensive in the Raman than in the IR spectrum.
Vibrational modes of the crystallization waters
Vibrational modes of H2O molecules under C2v symmetry are ν1 is the symmetric stretching vibration (νs), ν2 is the symmetric bending (δs), andν3 is the antisymmetric stretching vibration (νas). The total number of factor group modes due to the internal vibrations of water is 12. For three crystallographic types of water molecules that is equal to 36 vibrational degrees of freedom, external modes of H2O molecules under C2v symmetry: Rz = τ; Rx = ω; Ry = ρ. The total number of factor group modes due to the external vibrations of water is 24. For three crystallographic types of water molecules that equals 72 external degrees of freedom (36 hindered rotations and 36 hindered translations) (ESI Fig. 1), the degeneracies are lifted and all the internal and external mode bands are IR and Raman active. Both stretching modes of crystalline water are located in the higher wavenumber side of the band positions of NH stretching modes and appear around 3500 cm−1 as a shoulder. The bands of symmetric bending mode of water are expected to be located between 1600 and 1640 cm−1. However, in this range, there is a complex band system which contains not only the six types of symmetric OH2 deformation band (scissoring mode) but also the bands belong to the stretching mode of the non-coordinated C=O bonds of two kinds of carbonate ion and the eight kinds of an antisymmetric deformation mode of coordinated ammonia. The Raman spectra recorded with 532 nm excitation contains a complicated band system centered at 3495 cm−1 and assigned to the symmetric and antisymmetric stretching modes of crystalline water molecules in compound 1. One should expect a great extent of mode mixing as a result of the lowest possible symmetry of all units. A possible way to offer a more reasonable assignment (backed by experiment) would be to prepare isotopically substituted samples, where D2O substitutes water molecules. Water librations (hindered rotations) are expected to be highly affected by the H/D replacement (for pure modes, isotopic ratios close to 1.35 are expected) [39].
Lattice vibrations and combination bands
Over the assigned bands belonging to the cis-O2CoN4 skeleton, some lattice vibrations could also be found in the far-IR spectra at 56, 60, 76 and 96 cm−1. The Raman spectra recorded at 532 and 785 nm contains a weak lattice vibration at 114 cm−1 (ESI Figs. 3 and 4), while in the far-IR spectra (ESI Fig. 5), a wide shoulder system can be seen around this wavenumber value.

TG–DTG curve of compound 1 in air and N2 atmosphere
There is a multiple very wide band system located between 1700 and 2700 cm−1 and consists of overtones and combination bands of the species built the compound 1. The appearance of 2ν2 overtone (2082 and 1985) and ν1 + ν3 combination (2385 and 2347 cm−1) for carbonate ion, or ν1 + ν3(c) component overtone of sulfate ion (2082 cm−1) might be present in this band system, where c means the highest wavenumber component of the triply degenerate sulfate antisymmetric stretching mode [39].
UV spectra of compound 1
The carbonatoammine complexes of cobalt are low-spin complexes with d6(t 62g ) electron configuration and an electron excitation into the eg, orbital leads to t 52g eg configuration. The low-spin ground state in an octahedral field is 1A1g. Two-spin-allowed to singlet (1T1g and 1T2g) and two-spin-forbidden transitions to triplet states (3T1g and 3T2g) are possible when the triplet states lie at lower energy than the singlet ones. The UV–Vis spectrum of compound 1 can be seen in ESI Fig. 6 and Table 5.

DSC curves of compound 1 in air and N2
The purplish color of the compound 1 is related to the strongest spin-allowed transition (1T1g ← 1A1g) at 537 nm. The second principal band belongs to the other spin-allowed transition (1T2g ← 1A1g), while the two spin-forbidden transitions (3T1g ← 1A1g and 3T2g ← 1A1g) resulted in weak bands. The wavenumbers of these bands are sensitive to distortion of the octahedral symmetry, due to the hydrogen bonds influence on the shifting from the theoretically found values calculated by Sastri and Langford [41]. A CT band at 300 nm was assigned as an Lπ → Co(eg)-type ligand-to-metal-type (LMCT) electron transfer and confirms that the carbonate ion has more than 50% covalency in compound 1 [42].
Thermal studies on compound 1
The thermal decomposition processes of [CoCO3(NH3)4]SO4·3H2O (compound 1) have been studied since the late 1970s of the last century [21, 22]. The controversies in the results found in inert atmosphere and air make it highly desirable to prove the decomposition mechanism by modern, sophisticated techniques. Therefore, compound 1 was analyzed by simultaneous TG–DSC and coupled TG–MS measurements in both inert and oxidative atmosphere until 500 °C. The crystal hydrate [CoCO3(NH3)4]SO4·3H2O begins to lose its lattice water at about room temperature in both nitrogen and air (Fig. 5). This step is completed up to about 90 °C. Our results show that dehydration occurs at a significantly lower temperature (from r.t. to ~ 90 °C) than the published ones by Amigo [21] and Onodera [22] (from ~ 100 to ~ 190 °C). The dehydration process of complex 1 occurs in two overlapped steps, as seen from the DTG and DSC curves (Fig. 6). The peak temperature of water loss agrees with the result of Macikenas et al. (T = 59 °C), who confirmed the structure (identity) of their sample by single-crystal X-ray diffraction. The somewhat lower mass percent values (9.7% in N2 and ~ 9.0% in air) than the calculated for three water molecules (theoretical value is 10.31%) are also in accordance with spontaneous water lose even at room temperature.
By water loss, anhydrous [CoCO3(NH3)4]SO4 (compound 2) was formed, stable up to 221 °C onset, as it was found at isotherm heating of compound 1 at 120 °C in air [21]. There are no significant differences between the decomposition mechanism of compound 1 in inert and oxidative atmosphere up to 500 °C. In other words, environmental oxygen does not affect the decomposition features in this temperature range. To examine the effect of environmental oxygen on decomposition mechanism at higher temperatures and the final product, compound 1 was heated in both N2 and air atmosphere isothermally at 800 °C for 2 h.
Both Amigo and Onodera found ammonia and carbon dioxide evolution around 240 °C [21, 22], in one step, but only Onodera could detect N2 as an oxidation product of ammonia. We found 45.6% and 44.7% of mass loss in air and N2, respectively, which is somewhat more than the theoretical value (%). The reaction heat was found to be 437.49 kJ mol−1 and 507.18 kJ mol−1 in air and N2 atmosphere, respectively (DSC). These are consistent with formation of some residual ammonium sulfate detected by Onodera [22] and confirmed by us with IR studies on the decomposition intermediates formed at 300 °C. The difference in amount of ammonium sulfate in air and N2 causes some minor alteration in the measured reaction heats (Fig. 6). Decomposition intermediates to study the presence and absence of ammonium sulfate intermediate were made at 300 °C in both N2 and air atmosphere with and without prolonged heating. This intermediate was proved to be X-ray amorphous. The IR spectrum contained the characteristic bands of ammonium ions (δas = 1433 cm−1, δs = 1632 cm−1, ν1, ν3 3397 and 3263 cm−1, respectively, ν1 + ν5 at 3515 cm−1, ν2 + ν4 at 3178 cm−1, ν4 + ν6 = 1713 cm−1 and 2ν4 2916 cm−1 [43]) and strong sulfate bands (ν1 = 982 cm−1 (A), ν2 = 486 cm−1 (E), ν3 = 1118, 1063 cm−1 (F2) ν4 = 657,622,593 cm−1 (F2)) [39]. It has to be mentioned that small peaks of carbonate ion (substitutes the sulfate in the cobalt salts) may also observed at 833 and 1530 cm−1. The appearance of combination and overtone bands of ammonium ion might be attributed to the hindered rotation of ammonium ion as found in strong hydrogen-bond systems like (NH4)2SO4 [43, 44] (ESI Fig. 7). The bands of ammonium sulfate disappeared on prolonged heating (> 2 h). It is very probably that ammonium sulfate decomposes thermally and reacts with carbonate residues which causes deficit in Co/sulfate ratio and appearing excess of oxide as counter ion. The incompleteness of the cobalt(III) reduction in the main reaction steps (~ 240 °C) results in residual Co(III) content in the samples heated in air or inert atmosphere at 300 °C (5.3 and 2.8% in air and N2, respectively). The formula of compound 3 formed under oxidative (3a) and inert (3b) conditions at 300 °C was determined. In both cases, a kind of basic cobalt(II) sulfate was formed. The exact composition of the phases was found to be Co2O1,25+δ(SO4)0.75 (δ = the oxygen surplus due to presence of 5.3% of Co(III) ion (compound 3a) and Co2O1,14+δ(SO4)0.86 (δ = the oxygen surplus due to presence of 2.8% of Co(III) ion (compound 3b). It unambiguously showed the presence of a cobalt(III) reduction into cobalt(II) in aerial environment as well.

Thermal decomposition of compound 1 in the presence of oxygen
Compound 3a is oxidized at 719 °C and forms Co3O4. We could not detect complete oxidation of cobalt(II) in compound 3 to form cobaltyl sulfate (CoO)2SO4). As can be seen, besides the dehydration, the decomposition process is also endothermic independently of the atmosphere. However, the transformation of the unidentified minor intermediate is exothermic both in air and inert atmosphere. The small exothermic peaks were found at ~ 416 °C in air and ~ 473 °C in inert atmosphere. Mass change at these temperatures was practically not detectable. The differences might be attributed to the behavior of the minor unidentified intermediate toward oxygen and some other solid components in compound 3a in the lack of oxygen. Due to different behavior of this intermediate component in air and N2, to identify the possible role of oxygen/the lack of oxygen in these exothermic processes, a TG–DSC experiment in the presence of O2 was performed until 800 °C (Fig. 7).
The end product of decomposition was found to be Co3O4 in air, as stated by Amigo et al. [21]. However, in an inert atmosphere, a mixture of CoO and two allotropes (hcp and fcc cobalt) of metallic cobalt could be found in ca. 40:35:25 ratio (Fig. 8). The measurement has been repeatedly taken a high-temperature XRD as well when Al2O3 as a reference material was used to avoid the shifting in the diffractogram caused by the thermal expansion and phase transformation. Surprisingly, in this case, only crystalline CoO could be detected even at 800 °C or room temperature.

High-temperature XRD of the decomposition product of compound 1 in inert atmosphere measured at 450, 550 and 800 °C in the presence of Al2O3 a and XRD of the decomposition product heat treated isothermally in inert atmosphere at 800 °C b
The metallic cobalt formed in the redox decomposition step ~ 240 °C as one of the redox products. The hcp-Co:fcc-Co:CoO ratio was found to be 25:35:40. Oxidation of nanosize cobalt particles in air or O2 into CoO/Co3O4 might initiate the exoeffect at 415 °C. The cobalt oxidation temperature strongly depends on the size of cobalt particles and might be from room temperature until the temperature of red hot. In inert atmosphere, the lack of oxygen discloses this process and according to this, there is no exoeffect on the DSC curve at this temperature. The hexagonal-cubic phase transition temperature (hcp-fcc, 450–490 °C, its temperature strongly depends on impurity and grain size [45] of cobalt) coincides with the temperature of the exoeffect observed in inert atmosphere (473 °C), but the phase transition should be endothermic. Thus, this peak is attributed to the reaction of reactive nanosize cobalt (15 and 250 nm for hcp-Co and fcc-Co, respectively) particles with other components like CoO or Co2OSO4.
To examine the decomposition mechanism and the redox processes in inert and oxidative atmosphere, the evolved gases were analyzed by coupled TG–MS measurements (Figs. 9 and 10). Water evolution was found during the dehydration and the main decomposition steps. The appearance of water during the decomposition of the anhydrous [Co(NH3)4CO3]2SO4 confirms the existence of a redox reaction involving the ammonia. The only hydrogen source for water formation might be the ammonia. The water (m/z = 18, 17, 16 (H2O+, HO+ and O+) and ammonia curves (m/z = 17, 16, 15, 14 (NH +3 , NH +2 , NH+ and N+)) cannot be distinguished in the case of m/z = 17 and 16. However, the m/z = 18 peaks may belong only to water and the m/z = 15 or 14 may not belong to the water, but their source might be other nitrogen-containing parent ions as N +2 or N2O+. The m/z = 15 might also be double ionized N2O (N2O2+, m/z = 30/2)). We could not confirm Onodera [22] result about multistep ammonia losing and formation of a Co(III)-containing intermediate, ([Co(NH3)3CO3]2SO4, compound 4) at 200 °C. However, the N2 as a product of the redox reaction is verified by the appearance of m/z = 28 peak. Indeed, the CO2 fragmentation formed by ligand loss also results in CO+ as a fragment peak with the same m/z value. The situation is more complicated because the parent of CO+ is CO2, a typical carbonate decomposition product (m/z = 44) and a possible ammonia oxidation product (N2O) [46] have peak at this m/z value, and its possible fragments are NO+ (m/z = 30) or N +2 (m/z = 28).

TG–MS curves of compound 1 under argon atmosphere

TG–MS curves of decomposition products and their fragments on heating of compound 1 in air
Comparing the relative intensities of parent and their fragment ions for the H2O/NH3 and CO2/CO or N2O/N2/NO systems [47], it can roughly estimate the contribution of each fragmentation process to the intensities of the appeared fragment ions. For example, the high intensity of m/z = 17 comparing with m/z = 18 shows that the main component of m/z = 17 is the ammonia. A smaller contribution can be expected from the water fragmentation (m/z = 17, OH+) because the intensity ratio of m/z = 18 and m/z = 17 is ~ 4:1 in the case of water only.
Based on this, the intensity contribution of NH3 fragments (NH2) and oxygen from the water (O) can also be estimated, because the contribution of ammonia to this peak is ca. 80% related to the m/z = 17 (NH3). In comparison, in the case of water, it is negligible (~ 1% comparing with m/z = 18 intensity). The m/z = 16 peak may be due to any other oxygen-containing gases like CO2 or NOx compounds.
The N2O is a possible decomposition product of transition metal ammonia complexes with oxidizing ligands [1,2,3, 46, 48]. Unfortunately, it gives a peak on m/z = 44 like CO +2 . Their main fragments (CO+ or N +2 , respectively) also have the same m/z = 28; therefore, they cannot be distinguished. However, NO+, as a fragmentation product of N2O+, appears in the case of N2O only. But NO may also be a direct oxidation product of ammonia [1,2,3, 46]. Since the intensity value of m/z = 44 is comparable but smaller than the intensity of m/z = 30 (NO+), despite CO +2 contribution to the m/z = 44 peak, it suggests that NO+ is not a fragmentation product of N2O+ but a direct oxidation product of ammonia. The decomposition character of compound 1 in air confirmed that the same processes play a role in both atmospheres in the decomposition of 1 until 300 °C.
Summarizing the results of TG, DTG and TG–MS studies, combining with the IR and XRD results of intermediates and final decomposition products, the main thermal decomposition reactions of compound 1 are the following.
The reaction is reversible, endothermic, and the anhydrous salt has the same coordination sphere as the starting compound 1. The reaction proceeds between 40 and 60 °C.
The main decomposition reaction is an endothermic redox reaction between the ammonia ligands and cobalt(III) cation [49], both in an inert atmosphere and in air. The redox products are basic cobalt(II) sulfates, elementary nitrogen and water. Other products are carbon dioxide and ammonia. These are evolved together, but the decomposition probably starts with the ammonia oxidation process in the solid phase. A small amount of ammonium sulfate could be detected. However, the presence of ammonium nitrate as an oxidation product and N2O precursor could not be verified by FTIR measurement, because the nitrate bands coincide with the NH +4 bands around 1400 cm−1, and the N2O+ main peaks coincide with the CO +2 and CO+ fragment peaks formed from the leaving carbonate group. A minor amount of SO+ (m/z = 48) fragment could be detected from the SO2/SO3 parents [47]. It can cause the deficiency in sulfate/Co ratio (< 1:2) attributed to the ammonium sulfate gradual decomposes on prolonged isotherm heating or with increasing the decomposition temperature. Another ammonia oxidation product, NO, is also verified.
The basic cobalt sulfate, Co2OSO4, decomposes with the formation of CoO in an inert atmosphere but is oxidized into \({\text{Co}}^{{\rm II}} {\text{Co}}^{{\rm III}}_{2} {\text{O}}_{4} \left( {{\text{Co}}_{3} {\text{O}}_{4} } \right)\) in the air at 917 °C.
Characterization of compound 2
The compound 2 was prepared by thermal dehydration of compound 1 at 120 °C. The bands belonging to the cationic skeletal and ligand vibrations (ammonia and carbonate) could be assigned comparing the measured data with the results given for compound 1 (ESI Fig. 8). The bands belonging to crystallization water have disappeared. Numerous bands are overlapped with each other, e.g., δs(NH) and ν(C=O) stretching between 1650 and 1600 cm−1, and the antisymmetric and the symmetric C–O stretchings of carbonate ion are overlapped by antisymmetric S–O stretching observed at 1074 cm−1. The π(CO3) is mixed with ρ(NH3) at 833 cm−1. The νCoN(O) mode and the symmetric S–O deformation are mixed around 477 cm−1. The presence of two bands at 1516 and 1429 cm−1 appears only in the spectrum of compound 2; thus, these bands show some special (hydrogen bonding) interactions of ligands involving water of crystallization.
The spin-allowed and CT bands of the cation could be found for compound 2. The slight shifts of the wavenumbers shows that dehydration has no significant influence on the coordination of the complex cation. The two forbidden transitions cannot be observed. The appearance of the separated CT band around 300 nm in the spectrum of compound 2 instead of a shoulder observed in the spectra of compound 1 shows that the strong band covered by the CT band belongs to the water or water-bound species (~ 250 nm) (Water is bound by hydrogen bonds to sulfate [20] in compound 1.) The lattice parameters of compound 2 could not be determined by XRD due to insufficient crystallinity of the sample (ESI Fig. 9). The d values are given in ESI Table 1. The morphology of the compound 2 was studied by SEM, and the result can be seen in Fig. 11. SEM images indicate the [Co(NH3)4CO3]SO4 has a rugged surface. As can be seen, the crystallites possess irregular planar shape and aggregated in several micrometer-size.

SEM picture of [Co(NH3)4CO3]SO4
Characterization of CoO1.25+δCo(SO4)0.75 and CoO1.12+δCo(SO4)0.88 basic cobalt sulfates
The basic cobalt sulfates, CoO1.25+δCo(SO4)0.75 (compounds 3a) and CoO1.12+δCo(SO4)0.88 (compound 3b) were prepared as pink powders by heating of compound 1 at 300 C° for 2.5 h in air or N2 atmosphere for 2 h. Compounds 3a and 3b contain less sulfate ion than it was expected from the stoichiometry of compound 1 (sulfate:Co < 1:2). It might be attributed to the thermal decomposition of ammonium sulfate formed during the decomposition of compound 1 or 2 into basic cobalt sulfates. Compounds 3a and 3b have not been prepared and characterized yet. There are four different anhydrous basic cobalt(II) sulfates (2CoO·CoSO4, [50] 3CoO·CoSO4 [51], 4CoO·CoSO4 [52] and 5CoO·CoSO4 [50, 53] described in the literature. Two among them, the 2CoO·CoSO4 and the 4CoO·CoSO4, were only isolated in anhydrous form according to the given formula with heating the primarily formed hydrated basic salt at 415–525 and 292–295 °C, respectively [50, 52]. The other two references [50, 52] gave the CoO/SO3 ratio only for hydrated salts and did not isolate the anhydrous compounds. The cell parameters of compound 3a and 3b could not be determined because they were not enough crystalline to evaluate their diffractograms. Infrared spectrum of compound 3a shows the characteristic tetrahedral sulfate ion frequencies (νs = 984 cm−1, νas = 1107, 1057 cm−1, δas = 646, 566, 537 cm−1, sh, δs = 465 cm−1 (sh). The Co–O modes are assigned as a wide band system in the far-IR spectrum at 365 cm−1. The SEM picture can be seen in Fig. 12. Compound 3a occurs as rod-shaped blocks. Its surface is rather smooth, and it has a smaller grain size than the parent compound 2; however, it forms also bigger agglomerates.

SEM picture of Co2O1.25+δ (SO4)0.75
The photocatalytic activity of Co2O1.25+δ(SO4)0.75 (compound 3a) in the degradation of harmful dyes like Methyl orange (MO) and Congo red (CR) was studied at pH below and above the pKa values of the dyes. The methyl orange does not decompose photocatalytically with or without pH control, while the decomposition of Congo red was found to be reliable. The compound 3a has 53 m2 g−1 BET surface area. It shows adsorption capacity toward Congo red (30.7 mg CR g−1 of Co2O1.25(SO4)0.75). Under irradiation with a UV lamp for 240 min, the pseudo-first-order reaction rate constants were determined by linear regression of the data considering the Lagergren model at pH = 5.25. The apparent rate constant of the reaction (kapp) was obtained from the slope of the graph representation − ln(At/Ao) versus time and given in Table 6. The pseudo-first-order reaction model did not seem real in the case of the acid form of Congo red, probably due to the dissolution of the surface of catalysts in the acid changing the number and type of active sites. At pH = 5.25, however, the rate constant was found to be seven times higher than without catalyst (Table 6). Pseudo-second-order equations resulted in negative values of k2, which discloses this relationship from the evaluation of the measured data.
Characterization of final thermal decomposition products Co3O4 and CoO
The final decomposition/oxidation product (Co3O4) made at 750 °C from compound 1 in air has a small BET surface area (6 m2 g−1). It does not result in significant photocatalytic activity for the degradation of either Methyl orange or Congo red dyes. According to SEM images, Co3O4 shows microcrystalline structure (Figs. 13, 14).

SEM picture of Co3O4 crystals made in air at 750 °C

SEM picture of CoO formed from compound 1 in inert atmosphere at 800 °C
The final decomposition product (CoO) prepared at 800 °C from compound 1 under N2 atmosphere has a 1 m2 g−1 BET surface area. Its SEM pictures show different morphology comparing with the final decomposition product formed in air (Co3O4). It has large sheet-like structure; however, some small spherical particles also observed on the surface of the matrix, similar than the Co3O4 crystals in Fig. 13.
Conclusions
Di[κ1O,κ2O-carbonatotetraamminecobalt(III)] sulfate trihydrate, ([Co(NH3)4CO3]2SO4·3H2O dehydration by isothermic heating at 100 °C results in [Co(NH3)4CO3]2SO4 (compound 2) without alteration of the coordination sphere around. Compound 2 decomposes at ~ 240 °C in inert atmosphere giving a decomposition products consists of two modifications of nanosized metallic cobalt (hcp-15 nm, fcc-250 nm) and CoO (55 nm). The redox reaction results in N2 as an ammonia oxidation product. The decomposition intermediate is a cobalt(II) compound, Co2O1,14+δ(SO4)0.86 (δ = the oxygen surplus due to the presence of 2.8% of Co(III) ion). The same reaction in air atmosphere resulted in Co2O1.25+δ(SO4)0.75 (δ = the oxygen surplus due to presence of 5.3% of Co(III) ion (compound 3a). Compound 3a is oxidized in air at 793 °C into Co3O4. The compound 3a exhibits catalytic activity in photodegradation in Congo red. The photodegradation process follows pseudo-first-order kinetic (kapp = 1.0 and 7.0. at pH = 3.4 and 5.25, respectively).
References
Sajó IE, Kótai L, Keresztury G, Gács I, Pokol G, Kristóf J, Soptrayanov B, Petrusevski VM, Timpu D, Sharma PK. Studies on the chemistry of tetraamminezinc(II) dipermanganate [Zn(NH3)4](MnO4)2: Low-temperature synthesis of the manganese zinc oxide (ZnMn2O4) catalyst precursor. Helv Chim Acta. 2008;91(9):1646–8.
Kótai L, Banerji KK, Sajó IE, Kristóf J, Sreedhar B, Holly S, Keresztury G, Rockenbauer A. An unprecedented-type intramolecular redox reaction of solid tetraamminecopper(2+) bis(permanganate) ([Cu(NH3)4](MnO4)2)—a low-temperature synthesis of copper dimanganese tetraoxide-type (CuMn2O4) nanocrystalline catalyst precursors. Helv Chim Acta. 2002;85(8):2316–27.
Kótai L, Sajó IE, Jakab E, Keresztury G, Németh C, Gács I, Menyhárt A, Kristóf J, Hajba L, Petrusevski VM, Ivanovski D, Timpu D, Sharma PK. Studies on the chemistry of [Cd(NH3)4](MnO4)2. A low-temperature synthesis route of the CdMn2O4+x type NOx and CH3SH sensor precursors. Z Anorg Allgem Chem. 2012;638(1):177–86.
Kovács GB, May NV, Bombicz PA, Klébert S, Németh P, Menyhárd A, Novodárszki G, Petrusevski V, Franguelli FP, Magyari J, Béres K, Szilágyi IM, Kótai L. An unknown component of a selective and mild oxidant: structure and oxidative ability of a double salt-type complex having κ1O-coordinated permanganate anions and three-and four-fold coordinated silver cations. RSC Adv. 2019;9(49):28387–98.
Holló BB, Petruševski VM, Kovács GB, Franguelli FP, Farkas A, Menyhárd A, Lendvay G, Sajó IE, Nagy-Bereczki L, Pawar RP, Szilágyi IM, Bódis E, Kótai L. Thermal and spectroscopic studies on a double-salt-type pyridine—silver perchlorate complex having κ1-O coordinated perchlorate ions. J Therm Anal Calorim. 2019;138(2):1193–205.
Sajó IE, Kovács GB, Pasinszki T, Bombicz PA, May Z, Szilágyi IM, Jánosity A, Banerji KK, Kant R, Kótai L. The chemical identity of “[Ag(py)2]MnO4” organic solvent soluble oxidizing agent and new synthetic routes for the preparation of [Ag(py)n]XO4 (X = Mn, Cl, and Re, n = 2–4) complexes. J Coord Chem. 2018;71(16–18):2884–904.
Kótai L, Fodor J, Jakab E, Sajó IE, Szabó P, Lónyi F, Valyon J, Gács I, Argay G, Banerji KK. A thermally induced low-temperature intramolecular redox reaction of bis (pyridine) silver (I) permanganate and its hemipyridine solvate. Transit Met Chem. 2006;31(1):30–4.
Górska N, Mikuli E, Kótai L. Spectroscopic, structural and thermal characterization of crystalline [Cr(OC(NH2)2)6]X3 (X = ClO4, BF4 and Cl) complexes. Eur Chem Bull. 2014;3(5):474–81.
Mansouri M, Atashi H, Tabrizi FF, Mirzaei AA. Kinetics studies of nano-structured cobalt–manganese oxide catalysts in Fischer–Tropsch synthesis. J Ind Eng Chem. 2013;19(4):1177–83.
Mansouri M, Atashi H, Khalilipour MM, Setareshenas N, Shahraki F. Rate expression of Fischer–Tropsch synthesis over Co–Mn nanocatalyst by response surface methodology (RSM). J Korean Chem Soc. 2013;57(6):769–77.
Vortmann G. Zur Kenntniss der Kobaltammonium-Verbindungen. Ber Dtsch Chem Ges. 1877;10(2):1451–9.
Strock LW, McCutcheon TP. The crystalline form of some new cobaltammines. J Am Chem Soc. 1931;53(8):2852–66.
Udovenko VV, Gerasenkova AN. Monoethanolamine complexes of cobalt. Zh Neorg Khim. 1966;11(9):2066–9.
Hara R. Studies on the precipitation reactions of some metallic complex salts towards proteins. Yakugaku Zasshi. 1952;72(6):748–56.
Konoshita K. Feeding fungi with cobaltammine complex salts. Acta Phytochim. 1927;3:31–49.
Jaeger FM. Beiträge zur Krystalldiagnose der Kobaltverbindungen mit complexen Ionen. Z Kristallogr Cryst Mater. 1904;39(1–6):541–75.
Flint EE. Goniometric investigations. I. [Co(NH3)4CO3]2SO4·3H2O. Trudy Instituta Kristallografiya. Akademiya Nauk SSSR. 1947;3:11–2.
Strock LW. The crystallography and space group of carbonatotetraamminecobaltic sulfate. Z Kristallog Kristallgeom Krystallphys Kristallchemie. 1933;86:45–52.
Amigó JM, Torras CM. Constantes cristalográficas del [CoCO3(NH3)4]2SO43H2O. Acta Geol Hisp. 1973;8(5):165–6.
Macikenas D, Hazell RG, Christensen AN. X-ray crystallographic study of tetraamminecarbonatocobalt(III) sulfate trihydrate, [Co(NH3)4CO3]2SO4·3H2O. Acta Chem Scand. 1995;49(9):636–9.
Amigó JM, Garcia-González J, Miravitlles C. Thermal behaviour of [CoCO3(NH3)4]2SO43H2O. J Therm Anal Calorim. 1971;3(2):169–76.
Onodera S. Gas-chromatographic studies of the thermal decompositions of carbonato-pentaammine-, carbonatotetraammine-, and dicarbonato-diamminecobalt (III) complexes in the solid state. Bull Chem Soc Jpn. 1978;51(6):1889–90.
Vortmann G, Blasberg O. Zur Kenntniss der Kobaltoctaminsalze. Ber Dtsch Chem Ges. 1889;22(2):2648–55.
Benrath A, Kohlberg W. Hydrated salts of complex cobalt bases. Z Anorg Allg Chem. 1924;138:65–77.
Jörgensen SM. Zur Konstitution der Kobalt-, Chrom-und Rhodiumbasen. Z Anorg Chem. 1894;7(1):289–330.
Uspensky A, Tschibisoff K. Die Untersuchung einiger Ersatzreaktionen in der inneren Sphäre der Komplexverbindungen. Z Anorg Allg Chem. 1927;164(1):335–40.
Komarova AV, Pyartman AK, Kolobov NP, Mironov VE. Outer-spheric association of cobalt(III) carbonatotetraamine with anions. Zh Fiz Khim. 1974;48(4):1035–6.
Schwarz R, Tede K. Über die Photochemie der Komplexverbindungen (II). Ber Dtsch Chem Ges. 1927;60(B):63–9.
Ephraim F. Über die Löslichkeit von Kobaltiaken. (8. Beitrag zur Kenntnis der Löslichkeit.). Ber Dtsch Chem Ges. 1923;56(7):1530–42.
Erdey L. Titrimetric analysis. Budapest: Akadémia Kiadó; 1958 (in Hungarian).
Erdey L. Gravimetric analysis I-III. Budapest: Akadémia Kiadó; 1960 (in Hungarian).
Siebert H. Ultrarotspektren von Kobalt (III)-Komplexen mit Ammoniak und Resten von Sauerstoffsäuren als Liganden. Z Anorg Allg Chem. 1959;298(1–2):51–63.
Goldsmith JA, Hezel A, Ross SD. The skeletal vibrations of some cobalt (III) carbonato-, phosphato-and sulphato-complexes. Spectrochim Acta A. 1968;24(8):1139–47.
Goldsmith JA, Ross SD. Factors affecting the infra-red spectra of planar anions with D3h symmetry—IV the vibrational spectra of some complex carbonates in the region 4000–400 cm−1. Spectrochim Acta A. 1968;24(8):993–8.
Batsanov SS. Refraction of the hydrogen bond in inorganic compounds. Z Fiz Khim. 1960;34(1):68–77.
Dunell BA, Pachal MD, Ulrich SE. A study of thermal motion of ammine groups in carbonatotetramminecobalt (III) sulfate. Can J Chem. 1973;51(7):1107–8.
Ferraro JR. Low-frequency vibrations of inorganic and coordination compounds. New York: Plenum Press; 1971.
Fujita J, Martell AE, Nakamoto K. Infrared spectra of metal chelate compounds. VIII. Infrared spectra of Co(III) carbonato complexes. J Chem Phys. 1962;36(2):339–45.
Nakamoto K. Infrared and Raman spectra of inorganic and coordination compounds—part A and B. 6th ed. Hoboken: Wiley; 2009.
Sajó IE, Bakos LP, Szilágyi IM, Lendvay G, Magyari J, Mohai M, Szegedi A, Farkas A, Jánosity A, Klébert S, Kótai L. Unexpected sequential NH3/H2O solid/gas phase ligand exchange and quasi-intramolecular self-protonation yield [NH4Cu(OH)MoO4], a photocatalyst misidentified before as (NH4)2Cu (MoO4)2. Inorg Chem. 2018;57(21):13679–92.
Sastri VS, Langford CH. Electronic spectra of carbonato and oxalato ammine complexes of cobalt (III); average field model for singlet, triplet, and charge transfer bands. Can J Chem. 1969;47(22):4237–40.
Sastri VS. Studies on the disposition of carbonato group in cobalt (III) complexes. Inorg Chim Acta. 1972;6:264–6.
Schutte CJH, Heyns AM. Low-temperature studies. IV. The phase transitions of ammonium sulfate and ammonium-d4 sulfate; the nature of hydrogen bonding and the reorientation of the NX4+ ions. J Chem Phys. 1970;52(2):864–71.
Kótai L, Horváth T, Szentmihályi K, Keszler A. Evidence for quasi-intramolecular acid–base reactions in solutions of transition metal ammine complexes. Transit Met Chem. 2000;25:293–4.
Ray AE, Smith SR, Scofield JD. Study of the phase transformation of cobalt. J Phase Equilib. 1991;12(6):644–7.
Kótai L, Gács I, Sajó IE, Sharma PK, Banerji KK. Beliefs and facts in permanganate chemistry—an overview on the synthesis and the reactivity of simple and complex permanganates. Trend Inorg Chem. 2009;42(13):25–104.
Kocsis T, Magyari J, Sajó IE, Pasinszki T, Homonnay Z, Szilágyi IM, Farkas A, May Z, Effenberger H, Szakáll S, Pawar RP, Kótai L. Evidence of quasi-intramolecular redox reactions during thermal decomposition of ammonium hydroxodisulfitoferriate(III), (NH4)2[Fe(OH)(SO3)2]H2O. J Therm Anal Calorim. 2018;132(1):493–502.
Kristóf J, Horváth A, Szabó P. Simultaneous thermoanalytical investigations on the rapid decomposition of pentamminecobalt (III) complexes. J Therm Anal. 1990;36(3):1191–204.
Wendlandt WW, Smith JP. The thermal decomposition of metal complexes—VII A thermomagnetic study of the Co(III) → Co(II) reduction in cobalt(III) ammine complexes (1). J Inorg Nucl Chem. 1963;25(10):1267–72.
Dubler E, Oswald HR. New lower basic cobalt sulfate, Co3(OH)2(SO4)2·2H2O. Naturwissenschaften. 1969;56(6):327.
Pickering SU. The interaction of metallic sulphates and caustic alkalis. J Chem Soc Trans. 1907;91:1981–8.
Haberman J. Über einige basische Salze. Monatsch Chem. 1884;5:432–50.
Strömholm D. Studier öfver amorfa faellningar II. Om basiska salter af tvavarda metalloxider. Ark Kem Miner Geol. 1905/7;2(16):1–13.
Acknowledgements
Open access funding provided by Budapest University of Technology and Economics. The research within Project No. VEKOP-2.3.2-16-2017-00013 and GINOP-2.2.1-15-2017-00084 was supported by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. N. V. M. and P. B. are grateful for the Hungarian Scientific Research Found (K-124544 and K-115762). I. M. Szilágyi thanks for a János Bolyai Research Fellowship of the Hungarian Academy of Sciences and a ÚNKP-18-4-BME-238 grant supported by the New National Excellence Program of the Ministry of Human Capacities, Hungary. An NRDI K 124212 and an NRDI TNN_16 123631 grants are acknowledged. The research reported in this paper was supported by the Higher Education Excellence Program of the Ministry of Human Capacities in the frame of Nanotechnology and Materials Science research area of Budapest University of Technology (BME FIKP-NAT). B. Barta Holló acknowledges financial support of the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 451-03-68/2020-14/200125).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Franguelli, F.P., Barta-Holló, B., Petruševski, V.M. et al. Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products. J Therm Anal Calorim 145, 2907–2923 (2021). https://doi.org/10.1007/s10973-020-09991-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09991-3