
Abstract
The properties of alkali-activated materials (AAMs) depend on both the type of raw material used and their production procedure. This article presents an inexpensive and easily accessible method, based on using thermistors with a negative temperature coefficient, to analyse phenomena during the geopolymerisation process of AAMs. The described method enables prediction of the final physical and mechanical properties of tested materials and allows unambiguous determination of the quality of raw metakaolin materials in terms of their suitability for geopolymerisation processes and AAM production. This statement was proved by comparing AAMs formed based on metakaolin from three different sources. This article also describes the results of the mineralogical analysis, density, particle size distribution and morphology of the three metakaolins. In addition, the compression strength and FT-Raman spectroscopy of the AAM produced are described. Even though all materials were referred to as metakaolin, the results of this study showed that calcined materials can significantly differentiate the geopolymerisation process and final physical and mechanical properties of AAM.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alkali-activated materials and geopolymers now play an increasing role in industry, due to the ecology and economics of their production as well as a wide spectrum of possible applications. In addition, these materials could be designed to have superior properties compared to materials produced based on Portland cement, namely better resistance to acids and sulphate, better heat resistance, lower drying shrinkage and creep and higher strength [1].
The starting materials used for the synthesis of AAM and geopolymers are natural silicates (natural pozzolans) from kaolin or volcanic tuffs. The first step is metakaolin (Al2Si2O7) production as a result of kaolin (Al2Si2O5(OH)4) dehydroxylation. The main phenomena during this process are the transformation of octahedral Al into tetrahedral Al, structure amorphisation and retaining a 1:1 layer type. A study of the effect of the calcination temperature of kaolin on the properties of inorganic aluminosilicate polymers showed that geopolymers with optimal characteristics are obtained in a temperature range between 973 and 1073 K. Furthermore, it was found that the kaolin calcination heating high rate (K min−1) results in higher losses in the metakaolin, which are related to an increase in the residual kaolin. In the synthesis of geopolymers, the final properties are influenced by both the amorphous metakaolin phase and the residual kaolin content. Higher kaolin contents adversely affect the mechanical strength of the product. Therefore, to produce a geopolymer with high mechanical strength, kaolin calcination between 973 and 1073 K should be carried out at a low heating rate [2]. On the other hand, as demonstrated by differential scanning calorimetry (DSC), the calcination time has little effect on the reactivity of the material obtained during isothermal processing at 1023 K. The heat released during the polycondensation reaction for the raw material calcined for 2 h at 923 K was about 92% of the heat emitted as a result of the process for the material calcined at 1023 K for 4–6 h. The reactivity of the material after calcination for 2 h at 823 K was lower because the heat released was about 85% of the maximum registered for materials calcined at 1023 K [3].
The second step is the alkaline activation of metakaolin. The process of alkaline activation involves three types of reactions [3]. The first type leads to the dissolution of metakaolin into silicate monomers and aluminate monomers. This phenomenon has a strong exothermic character. The second type includes the polymerisation processes of these monomers into aluminosilicate oligomers, which immediately polymerise into small geopolymeric fragments or ‘proto-zeolitic nuclei’. They are, however, thermodynamically metastable and incompletely cross-linked. Therefore, the third stage involves their combination into larger molecules, i.e. into aluminosilicate inorganic polymer gels and crystallised phases, which allows obtaining a material with stable properties [4]. One major factor affecting the composition of alkali-activated material binding phases is the calcium content; N–A–S–H, C–(N)–A–S–H and C–A–S–H gels are formed in low-, intermediate- and high-calcium systems, respectively. However, in high-calcium systems, the main binding phase is crystalline C–S–H [5]. The reactivity of metakaolin also depends on the Al2O3 and SiO2 content; the higher the Al2O3 and SiO2 content, the more reactive the metakaolin [6]. Moreover, altering the Si/Al molar ratio allows the synthesis of materials with different structures as the aluminium atoms cross-link chains of SiO4 and MAlO4 tetrahedra (where M is a monovalent cation, typically Na+ or K+). The polymer formation rate is also influenced by alkali concentration. An analysis of DSC curves showed that the polymerisation reaction (starting after the dissolution process) increased with the NaOH solution concentration [3]. Furthermore, various parameters, such as the material’s specific surface area, initial solids content, purity and reaction temperature, result in the creation of different structures and networks [6,7,8,9,10,11,12].
Finally, the materials obtained need to be cured at ambient (approximately 298 K) or elevated (313–353 K) temperatures depending on precursors, mix design, relative humidity, etc. Generally, heat curing accelerates the early strength development of materials [5]; however, curing at advanced elevated temperatures for a prolonged period causes specimen deterioration due to the destabilisation of the silicate–Si–O–Al–O–bond.
Due to the depletion of natural metakaolin resources and aspects related to reasonably managing post-process waste (with respect to environmental protection), research is being carried out on using other aluminosilicate raw materials such as fly ash, blast furnace slag, volcanic ash [13,14,15,16,17] and post-mining waste [18,19,20]. The different sources of materials used to produce AAMs and geopolymers generate a very common production problem; their diverse and unique chemical composition results in unpredictable product physical and mechanical properties. There are also no simple and inexpensive test methods for the unambiguous quality determination of such raw materials in terms of their suitability for geopolymerisation processes or for producing AAMs. Nevertheless, this article presents a new method using negative temperature coefficient (NTC) thermistors that allows measurement of the thermal effects occurring during the polymerisation process of materials from different sources and estimation of the mechanical properties of these materials.
Materials and methods
Materials
Three types of metakaolins (M1, M2 and M3), from two different commercial companies (Astra, Poland, and the Sedlecký Kaolin A.S., Czech Republic), were used for the test. For the M1 and M2 materials, the procedure for their production was similar and based on calcination in rotary kilns, while the calcination of M3 metakaolin took place in flash-type furnaces.
Metakaolin characterisation methods
In order to determine the morphology of materials originating from different sources (M1–M3), tests were carried out on a JEOL JSM 820 Scanning Electron Microscope (SEM) with an energy-dispersive spectrometer (EDS) IXRF Systems Model 500 Digital Processing. The metakaolin particles were dried to constant mass, attached to coal tape and covered with a thin layer of gold using the JEOL JEE-4X vacuum evaporator. The mineralogical phases present in the sample were determined using X-ray diffraction analyses (Rigaku X-ray diffractometer) applying the following settings: CuKα radiation, a reflective graphite monochromator, 45 kV lamp voltage, 200 A lamp current, 0.05 2θ step and 1 s counting time per step. For phase identification, measured values of the lattice parameters were analysed against the ICDD (International Centre for Diffraction Data 2016) catalogue in the Xrayan program. The laser diffraction particle size analysis of metakaolin was performed in accordance with ISO 13320 using an Analysette 22 (Fritsch). The specific surface areas were determined with the BET (Brunauer–Emmett–Teller) method of multilayered nitrogen (99.999% purity) adsorption using an Autosorb iQ. Material density was measured using a helium Pycnomatic ATC.
Geopolymer preparation
To produce geopolymer materials, metakaolin and a sand mixture (1:1) were used. A NaOH solution (10 M, Sigma-Aldrich) with an aqueous sodium silicate solution (R-145, molar ratio 2.5, density 1.45 g cm−3) was applied for the activation process. The solid-to-liquid ratio was 2. The metakaolin, sand and alkaline solution were mixed for 15 min to form a homogeneous paste in a low-speed mixing machine. The prepared mixtures (M1–M3) were moved into a cylindrical form of the same volume, in accordance with EN 13791.
Geopolymerisation process characterisation methods
Standard negative temperature coefficient thermistors (NTCLE100E3, Vishay) were used to build the probe system. The thermistors consisted of a chip with two solid copper–tin-plated leads. Thermistors with resistance Rref = R25 = 10,000 Ω were used in the tests. The B25/85 value was 3977 K. The thermistors were protected against direct contact with the geopolymer mortar by polyethylene heat-shrink tubing and silicone resistant to elevated temperatures (up to 463 K). The correctness of thermistor measurements was checked each time in distilled water. A measuring element based on the NTC thermistor system was placed in the centre of the cylindrical samples (M1–M3) for measuring the thermal effects related to the chemical reaction that takes place during the polymerisation process. Due to the alkaline pH of the geopolymer mass, a new thermistor was used for each sample. The changes of resistance value were registered using an ATLAS 0531 impedance analyser. The measurements were performed over 24 h in a furnace at 348 K (geopolymerisation process). The samples were then moved to ambient temperature for 48 h, after which the next round of measurements was completed. Measurements were performed during the 24 h in the furnace at 348 K (using a geopolymer as a reference sample). Based on current–voltage data obtained during the measurements, the thermal effects were calculated using the Steinhart–Hart law [21]. The characteristic Steinhart–Hart coefficients are based on temperature-resistance tables for given thermistors and functions (extended Steinhart–Hart polynomial) allowing the conversion of temperatures to resistances and vice versa:
where T is the temperature in Kelvin; A1, B1, C1 and D1 are the Steinhart–Hart coefficients, which vary depending on the thermistor type and model and the temperature range of interest; and R is the resistance, in ohms, at T.
A Vicat needle apparatus was used to determine the initial setting time of mortar geopolymer pastes during polymerisation in the furnace at 348 K, according to PN-EN 196-3:2016-12. The beginning of the setting process was determined by the penetration behaviour of a steel needle into a geopolymer paste sample at a 300 s frequency.
Geopolymer characterisation methods
Spectroscopy analyses of geopolymers after 28 days of curing were carried out using an FT-Raman Nicolet NXR 9650 (Spectro-Lab) equipped with a Nd:YAG3 + laser with a 1064 nm wavelength. For each material, a minimum of 3 spots were measured (50 µm).
Compressive strength tests were carried out according to EN 13791 on cylindrical forms (Ø = 50 mm; h = 100 mm). The test was performed after 28 days of curing in 50% humidity and about 294 K on a Matest (Italy) testing machine, model 3000 kN.
Results and discussion
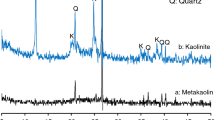
Figure 1 presents the results of XRD analyses of the three types of metakaolin used in the tests. There were significant differences in the phase composition of materials from different sources. Mullite, quartz, kaolinite, anatase, illite and opal phases were identified in descending order during analysis. M3 is characterised by a very high content of mullite phase, as evidenced by an abundant number of peaks on the X-ray diffraction pattern. The mullite phase was not found in M1 and in M2’s raw material; the mullite contribution was minor. After thermally treating kaolin at 873–973 K, the characteristic peaks for kaolinite (2θ 12.41°, 20.21° and 25.49°) should disappear, while peaks assigned to quartz (2θ 21.22° and 27.45°) should remain unchanged [22]. The occurrence of a kaolinite (Al2Si2O5(OH)4) phase, which indicates the incomplete transformation of kaolinite into metakaolinite, was noted for M2. A larger amount of quartz (SiO2) was found in M1 than in raw material M2, whereas in M3, the amount of quartz was insignificant. The comparison between materials also revealed certain changes regarding the halo peak representing the amorphous phase. A larger halo peak with 2θ between 18° and 38° was found for metakaolins M2 and M3, than for M1.

XRD patterns of three types of metakaolins M1, M2 and M3
Taking into consideration all the above results, one can assume that materials M1 and, to a lesser degree, M2 will be most useful in geopolymerisation processes. M2, due to the presence of residual kaolinite, was probably calcined at a low temperature or the calcination time was too short. At low temperatures (below 973 K), calcination produces residual kaolinite clays that make the product less reactive, whereas at higher temperatures (above 1123 K), re-crystallisation occurs, leading to the formation of non-pozzolanic materials such as spinel, silica and mullite [23]. In contrast, M3 was probably calcined at temperatures that were much higher than required, as evidenced by the presence of a mullite phase. As was proven in a detailed thermodynamics study of the kaolinite–mullite transformation, which was made at 1223–1773 K, the most stable transformation in this temperature range was the one that yields mullite rather than γ-alumina [24].
Representative morphology of metakaolins M1–M3 is shown in Fig. 2. An irregular shape was observed for all metakaolin particles. However, a particle population with the smallest size and the most angular and salient shape was found for the M3 metakaolin. The laser diffraction particle size analysis showed that the mode of particle size for M1 and M2 was about 23 and 26 μm, respectively, whereas M3 had one order of magnitude smaller particles, about 2 μm (Table 1). Nonetheless, particles of M1 and M2 were more homogeneous in the size since the particle size distribution width was reduced for M1 and M2 in comparison with M3 (Fig. 3); and the span values were 0.6, 1.1 and 2.8 for M1, M2 and M3, respectively (Table 1). It should be also noted that the particles (Fig. 2) were combined into aggregates; thus, their size measurements may not reflect the reactivity of the material during polymerisation processes. This statement was strengthened by the BET specific surface area measurement. Based on the particle size results (above), the M3 material should have a larger BET surface compared to M2 [25] (only M2 and M3 were compared due to the similar particle size distribution of M1 and M2). However, M3 metakaolin has nearly three times less specific surface area (4.5 m2 g−1) despite the lower particles size, compared to the M2 metakaolin specific surface area (13.0 m2 g−1). Altogether, the results show that metakaolins can produce significant differences in the physical and chemical properties, depending on the origin source and calcination process. It can be assumed that the effect of particle size can be related to the calcination time, which was too long for M3, thus the formation of sinters with low porosity and higher density (Table 1) resulting in a small surface development. Earlier studies proved this dependence, as the instantaneous thermal treatment generated vitrified particles with perfect roundness, whereas the prolonged calcination sequence generated a particle population with an angular and salient shape [25]. Additionally, the high quartz content present in M1 (and then in M2) can induce an effect in which rounded particles are minimally or not at all affected by the calcination process and thus maintain the reactivity properties of metakaolins more suitable for a successful polymerisation process [25]. This result is in agreement with the results from SEM analysis; M1 and M2 tended to have more rounded and smoothed particles in some classes. Further interpretation involves the existing amount of illite in M1 and M2 (Fig. 1), which promotes elongation and enlargement of particles [25].

Morphology of three types of metakaolins M1, M2 and M3

Particle size distribution of three types of metakaolins M1, M2 and M3
The initial setting time of the geopolymer mortar, measured with the Vicat needle apparatus at 348 K, depended on the type of metakaolin used. M1 and M2 were characterised by similar bonding times of 2250 ± s and 3000 ± s, respectively, while samples prepared from raw material M3 required almost twice the bonding time, 5100 ± s. Relatively long bonding times (40–80 min) could be a result of low calcium oxides content (Table 2), as they significantly accelerate the bonding processes [26]. Calcium-rich materials accelerate the setting of alkali-activated fly ash-based materials where additional calcium silicate hydrate (C–S–H) coexists with geopolymer products [27]. The results are also in agreement with an effect of metakaolin material particle size. The setting time was longest for M3, with the smallest particles. Similarly, the particles of cement obtained from a 140 mesh (105 µm) showed the longest hardening time, 22 min, which was different when compared to 60 mesh (250 µm, 17 min) and 100 mesh (149 µm, 17 min) [28].
Figures 4–6 present the thermal effects recorded with the NTC thermistor system, during the geopolymerisation process as well as 48 h after geopolymer curing (reference sample) for individual materials M1 (Fig. 4), M2 (Fig. 5) and M3 (Fig. 6). Several characteristic effects can be determined for each material: the maximum value of the thermal effects, the time when thermal effect reaches the highest value and the end-set time of thermal effects (Table 3). The highest value, 363 K, for the thermal effects was recorded during the test of M1 (Fig. 4, Table 3). The M2 raw material was characterised by lower value, 357 K (Fig. 5, Table 3), while the lowest one, 348 K, was noted for M3 (Fig. 6, Table 3). The time distribution of thermal effects was negatively related to temperature; the higher the temperature, the shorter the time of both the maximum effects and their extinction. Similarly, a subtraction performed between the temperature curve recorded for the geopolymerisation process and for the corresponding formed geopolymer, showed that raw material M1 is characterised by the most intense thermal effects with the shortest duration of about 6 h. In comparison with M1, the amplitude of the exothermic effect for M2 and M3 was lower (about 30% and 80%, respectively) and took longer (about 7 and 10 h, respectively). It is believed that the exothermic effects result mainly from dissolution of the solid metakaolin in a strongly alkaline aqueous solution and the breaking down of metakaolin particles together with the formation of oligomeric species in the aqueous phase in combination with the formation of alumina/silica–hydroxy species. The polymerisation/condensation reactions can be attributed to a small contribution of heat evolution, which implies that the process of network consolidation is rather thermally steady state [29]. The results are fully consistent with the setting time data measured by the Vicat method since M1 and M2 were characterised by shorter bonding times than M3. However, it should be noted that too high a temperature for such reactions, approaching the boiling temperature of water, is adverse to the formation of geopolymer because it can affect mechanical properties as an effect of abundant porosity. At 363 K, the exothermic reactions would cause more rapid water loss than at lower temperatures, leading to the formation of micro-cavities that cause an increase in porosity during network formation (Fig. 7a–c). The temperatures control the porosity development between G1 and G2. The reduced porosity affected the highest compressive strength, which was measured after 28 days of curing for G2 (Table 4) in comparison with improved porosity and lowered values of compressive strength recorded for G1 geopolymer. On the other hand, geopolymer G3 had more than ten times lower values of compressive strength despite G3’s porosity being comparable to G1 and G2 and having the smallest particles size of metakaolin M3. However, this effect was a result of the chemical composition of metakaolin M3 (presence of mullite phase), the angular shape and inhomogeneous distribution of its particles, the lowest specific surface area and the lowest reactivity (reduced thermal effects recorded with NTC thermistor and longer bonding time measured using the Vicat method).

Thermal effects recorded with the NTC thermistor system during the geopolymerisation process (M1, red curve) and 48 h after curing (G1, reference sample, blue curve), and the subtraction performed between temperature curves M1 and G1 (green curve). (Color figure online)

Thermal effects recorded with the NTC thermistor system during the geopolymerisation process (M2, red curve) and 48 h after curing (G2, reference sample, blue curve), and the subtraction performed between temperature curves M2 and G2 (green curve). (Color figure online)

Thermal effects recorded with the NTC thermistor system during the geopolymerisation process (M3, red curve) and 48 h after curing (G3, reference sample, blue curve), and the subtraction performed between temperature curves M3 and G3 (green curve). (Color figure online)

Representative macrostructure of geopolymers G1 (a), G2 (b) and G3 (c). Representative microstructure of geopolymers G1 (d), G2 (e) and G3 (f) prepared for FT-Raman analysis. The area of spectroscopy analysis was marked with a red square. (Color figure online)
Figure 7d–f presents representative microstructures of the geopolymers with marked areas of FT-Raman spectroscopy analysis. The recorded spectra for G1 and G2 are similar, while the spectrum of G3 was significantly different (Fig. 8). For geopolymers made of M1 or M2 metakaolin, multiple bondings in the 460–465 cm−1 range indicate the presence of Si–O–Si, Si–O–Al or O–Si–O bending vibrations, whereas in the 550–1159 cm−1 region, Si–O–Si or Si–O–Al symmetrical stretching vibrations were found [30,31,32]. In addition, these materials exhibit weak effects at 3419 cm−1, which correspond to O–H stretching vibrations. The intensity of the G2 peaks was significantly lower compared to G1, which can suggest a greater cross-linking of the aluminosilicate bonds in G2. This effect provides an explanation of the compressive strength results presented in Table 4. In G3, C–O, N=C=S, C≡C, C=N, O–H and H–O–H stretching vibrations predominated [32,33,34]. Moreover, for this sample, there are no effects from aluminosilicate bonds and this effect can be related to the lowest compressive strength recorded for G3 (Table 4).

FT-Raman spectra of three types of geopolymers G1, G2 and G3
Conclusions
Based on the results, the applicability of using an NTC thermistor probe system as a low-cost method for analysing phenomena occurring during geopolymerisation processes was confirmed. The presented method, together with other analyses, allowed unambiguous determination of the quality of raw metakaolin materials in terms of their suitability for geopolymerisation processes and producing AAMs. Even though all materials were referred to as metakaolin, the results of this study showed that calcined materials can significantly differentiate the geopolymerisation process and final physical and mechanical properties of AAMs. The NTC thermistor system allowed prediction of the mechanical properties of produced materials, e.g. their compressive strengths.
References
Škvára F. Alkali activated materials or geopolymers. Ceram Silik. 2007;51:173–7.
Kenne Diffo BB, Elimbia A, Cyr M, Dika Manga J, Tchakoute Kouamoa H. Effect of the rate of calcination of kaolin on the properties of metakaolin-based geopolymers. J Asian Ceram Soc. 2015;3:130–8.
Granizo ML, Blanco MT. Alkaline activation of metakaolin an isothermal conduction calorimetry study. J Therm Anal Calorim. 1998;52:957–65.
Zhang Z, Wang H, Provis JL, Bullen F, Reid A, Zhu Y. Quantitative kinetic and structural analysis of geopolymers. Part 1. The activation of metakaolin with sodium hydroxide. Thermochim Acta. 2012;539:23–33.
Luukkonen T, Abdollahnejad Z, Yliniemi J, Kinnunena P, Illikainen M. One-part alkali-activated materials: a review. Cement Concrete Res. 2018;103:21–34.
Tongbo S, Bin W, Lijun Z, Zhifeng C. Meta-kaolin for high performance concrete. In: Scrivener K, Favier A, editors. Calcined clays for sustainable concrete. RILEM Bookseries, vol 10;2015.
Hebda M, Laska M, Szechyńska-Hebda M. Application of a device used for observation of controlled thermal processes in a furnace: examples of delubrication, oxidation, melting, pyrolysis, and combustion. J Therm Anal Calorim. 2013;114(3):1099–109.
Grela A, Łach M, Bajda T, Mikuła J, Hebda M. Characterization of the products obtained from alkaline conversion of tuff and metakaolin. J Therm Anal Calorim. 2018;133(1):217–26.
Zhang MH, Malhotra VM. Characteristics of a thermally activated aluminosilicate pozzolanic material and its use in concrete. Cem Concr Res. 1995;25:1713–25.
San Nicolas R, Cyr M, Escadeillas G. Characteristics and applications of flash metakaolins. Appl Clay Sci. 2013;83–84:253–62.
Alonso S, Palomo A. Alkaline activation of metakaolin and calcium hydroxide mixtures: influence of temperature, activator concentration and solids ratio. Mater Lett. 2001;47:55–62.
Mohd Salahuddin MB, Norkhairunnisa M, Mustapha F. A review on thermophysical evaluation of alkali activated geopolymers. Ceram Int. 2015;41:4273–81.
Davidovits J. Geopolymer: chemistry and applications. 4th ed. Saint-Quentin: Geopolymer Institute; 2015.
Provis L, van Deventer JSJ. Geopolymers: structure, processing, properties and industrial applications. Sawston: Woodhead Publishing; 2009.
Łach M, Korniejenko K, Mikuła J. Thermal insulation and thermally resistant materials made of geopolymer foams. Procedia Eng. 2016;151:410–6.
Grela A, Łach M, Mikuła J, Hebda M. Thermal analysis of the products of alkali activation of fly ash from CFB boilers. J Therm Anal Calorim. 2016;124(3):1609–21.
Mikuła J, Łach M. Geopolymers—a new environment friendly alternative to concrete based on Portland cement. Part 2—examples of implementations and test results. In: Mikuła J, editor. Ecological solutions in the field of production: modern composite materials friendly to the environment. Cracow: Cracow University of Technology; 2014. p. 13–32.
Cheng Y, Hongqiang M, Hongyu C, Jiaxin W, Jing S, Zonghui L, Mingkai Y. Preparation and characterization of coal gangue geopolymers. Constr Build Mater. 2018;187:318–26.
Huang G, Ji Y, Li J, Hou Z, Dong Z. Improving strength of calcinated coal gangue geopolymer mortars via increasing calcium content. Constr Build Mater. 2018;166:760–8.
Geng J, Zhou M, Li Y, Chen Y, Han Y, Wan S, Zhou X, Hou H. Comparison of red mud and coal gangue blended geopolymers synthesized through thermal activation and mechanical grinding preactivation. Constr Build Mater. 2017;153:185–92.
Steinhart JS, Hart SR. Calibration curves for thermistors. Deep Sea Res Oceanogr Abstr. 1968;15:497–503.
Biljana R, Ilić A, Mitrović L, Miličić R. Thermal treatment of kaolin clay to obtain metakaolin. Hem Ind. 2010;64(4):351–6.
Ayininuola GM, Adekitan OA. Characterization of Ajebo Kaolinite Clay for production of natural pozzolan. Int J Civ Environ Struct Constr Arch Eng. 2016;10(9):1212–9.
Schieltz NC. Thermodynamics of the various high temperature transformations of kaolinite clay. Clay Miner. 1964;13(1):419–28.
Cassagnabère F, Diederich P, Mouret M, Escadeillas G, Lachemi M. Impact of metakaolin characteristics on the rheological properties of mortar in the fresh state. Cement Concrete Compos. 2013;37(1):95–107.
Tanomaru-Filho M, Garcia AC, Bosso-Martelo R, Berbert FLCV, Reis JMSN, Guerreiro-Tanomaru JM. Influence of addition of calcium oxide on physicochemical properties of Portland cement with zirconium or niobium oxide. J Conserv Dent. 2015;18(2):105–8.
Chindaprasirt P, Phoo-ngernkham T, Hanjitsuwan S, Horpibulsuk S, Poowancum A, Injorhor B. Effect of calcium-rich compounds on setting time and strength development of alkali-activated fly ash cured at ambient temperature. Case Stud Constr Mater. 2018;9:e00198.
Pecora JD, Silva RG, Savioli RN, Vansan LP. Effect of particle size of grossman’s cement powder on setting time. Rev Odontol Univ São Paulo. 1998;12(1). http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-06631998000100002&lng=en&nrm=iso.
Muñiz-Villarreala MS, Manzano-Ramíreza A, Sampieri-Bulbarelaa S, Gasca-Tiradoa JR, Reyes-Araizaa JL, Rubio-Ávalosa JC, Pérez-Buenob JJ, Apatigac LM, Zaldivar-Cadenad A, Amigó-Borrás V. The effect of temperature on the geopolymerization process of a metakaolin-based geopolymer. Mater Lett. 2011;65(6):995–8.
Nkuna CN, Oboirien BO, Sadiku ER, Lekitima J. A comparative study of geopolymers synthesized from OXY-combustion and chemical looping combustion bottom ashes. Constr Build Mater. 2017;136:246–55.
Jang JG, Lee HK. Effect of fly ash characteristics on delayed high-strength development of geopolymers. Constr Build Mater. 2016;102:260–9.
Song M, Jiaping L, Wei L, Yaocheng W, Jianzhong L, Liang S, Qian J. Property and microstructure of aluminosilicate inorganic coating for concrete: role of water to solid ratio. Constr Build Mater. 2017;148:846–56.
Król M, Minkiewicz J, Mozgawa W. IR spectroscopy studies of zeolites in geopolymeric materials derived from kaolinite. J Mol Struct. 2016;1126:200–6.
Yaseri S, Hajiaghaei G, Mohammadi F, Mahdikhani M, Farokhzad R. The role of synthesis parameters on the workability, setting and strength properties of binary binder based geopolymer paste. Constr Build Mater. 2017;157:534–45.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mierzwiński, D., Łach, M., Hebda, M. et al. Thermal phenomena of alkali-activated metakaolin studied with a negative temperature coefficient system. J Therm Anal Calorim 138, 4167–4175 (2019). https://doi.org/10.1007/s10973-019-08471-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08471-7