
Abstract
This paper studies hydration stages and phase transformation mechanism of the Sr2+-doped calcium zirconium aluminate cement at room temperature. Features of different hydration stages of this cement paste are identified by X-ray diffraction, scanning electron microscopy, differential thermal analysis, thermogravimetric analysis, evolved gas analysis and heat evolution test. It was found that the partial isovalent substitution of Ca2+ for Sr2+ such as Ca7−xSrxZrAl6O18, with x = 0.3, 0.6, 1.0, reduced the hydraulic reactivity of Ca7ZrAl6O18 phase. The course of hydration of mixed oxides of the type 6CaO·SrO·3Al2O3·ZrO2 documented using microcalorimetry was supported by investigations of the solid hydration products. Research showed that the hexagonal hydrates were stable at early and middle curing ages, an indication that including Sr in solid solution could effectively inhibit the conversion from both C4AH13–19 and C2AH8 to C3AH6 and AH3. Calcium monocarboaluminate \({\text{C}}_{4} {\text{A}\bar{\text{C}}\text{H}}_{11}\) phases were also found in this system due to the carbonation process of C4AH13 phase. Substitution of Ca2+ ions by Sr2+ ions in hexagonal calcium aluminate hydrates causes structural disorder and then contributes to the broadening of lines in the powder X-ray diffraction patterns of layered structures of metastable hydrates. At later curing ages, the formation of two hydrogarnet phases, one Sr-rich (C,Sr)3AH6 and the other Ca-rich (C,Sr)3AH6, was proved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Calcium aluminates, i.e. calcium monoaluminate (CaAl2O4, CA), calcium dialuminate (CaAl4O7, CA2) and dodeca-calcium hepta-aluminate or mayenite (Ca12Al14O33, C12A7), are the most important constituents of hydraulic calcium aluminate cements (CACs) [1, 2]. Calcium zirconium aluminate (Ca7ZrAl6O18, C7A3Z) is considered to be only a chemical compound containing zirconium (Zr) in CaO–Al2O3–ZrO2 system that exhibits hydraulic reactivity similar to tricalcium aluminate (Ca3Al2O6, C3A) [3,4,5]. It is also well known that the hydraulic reactivity of these phases increases with the calcium content of the phase (CaO/Al2O3 molar ratio), and therefore, C7A3Z shows higher reactivity than mineral phases of CACs. The formation and dehydration of a number of C–A–H-like phases of hydrated Ca7ZrAl6O18 were investigated in previous works [6,7,8,9,10,11,12].
The hydration process of the calcium aluminate cement produces similarly the various forms of hydrated calcium aluminate and gibbsite. Dissolution of the anhydrous phases causes a significant increase in concentration of Ca2+, Al(OH) −4 and OH− ions [13]. The precipitation and growth of the hydrates takes place through the solution. When supersaturation is reached, calcium and aluminium ions can combine in variable ratios to precipitate different hydrates CAH10, C2AH8, C4AH19, C4AH13, C3AH6 and AH3 (C ≡ CaO, A ≡ Al2O3, H ≡ H2O). These processes proceed with time and exhibit the temperature dependence [14]. The C2AH8 and C4AH19 belong to the broad AFm phases with the general formula [Ca2(Al,Fe)(OH)6]·X·xH2O, where X is one formula unit of a singly charged anion (e.g. OH−) or half a formula unit of a doubly charged anion (e.g. CO 2−3 ). These crystals have a layered structure consisting of positively charged [Ca2Al(OH)6]+ sheets with Al(OH) −4 or OH−, together with H2O in the interlayer region. C4AH19 contains a complete additional layer of H2O between the principle layers. C4AH19 can lose part of its interlayer water molecules to form C4AH13 at relative humidity below 88%. The initially formed metastable hexagonal hydrates are generally transformed into stable C3AH6 belonging to the regular system. The factors controlling transformation are: time, temperature, water content and CO2 content in the system. In the presence of atmospheric CO2, part of the interlayer OH− can be replaced by CO 2−3 via interlayer exchange to form two representative carbon-containing AFm phases: stable monocarboaluminate C3A·CaCO3·11H2O (\({\text{C}}_{4} {\text{A}\bar{\text{C}}\text{H}}_{11} ;\;{\bar{\text{C}}} = {\text{CO}}_{2}\)), which contains only CO 2−3 anions in the X position, and unstable hemicarboaluminate C3A·0.5CaCO3·0.5Ca(OH)2·11.5H2O (\({\text{C}}_{4} {\text{A}}\bar{\text{C}}_{0.5} {\text{H}}_{12}\)), which contains both OH− and CO 2−3 anions in the X position of hexagonal hydrates [15].
Due to a poor crystallinity of the hydration products formed at early age hydration and low water-to-cement ratio (w/c) or overlapping peaks due to hydrates with distinct but closely related crystallographic characteristics from a complex XRD profile, the methods of combined thermal analysis (DTA–TG–EGA) are promising ones regarding their qualitative and quantitative characterization. Previous works [16,17,18,19,20,21,22,23,24,25,26,27] indicated various decomposition temperatures of Ca–Al hydrates, obtained by thermal analysis methods. According to both Fleisher [16] and Hill [17], C4AH13–19 phase decomposes in the temperature range 200–280 °C or at a temperature of 250 °C, respectively. \({\text{C}}_{4} {\text{A}}\bar{\text{C}}{\text{H}}_{11}\) and C2AH8 do indeed give DTA peaks in the same temperature range 160–200 °C [18]. George [19] reports an endothermic peak due to dehydration of C2AH8 at 170 °C. Various authors [9, 20,21,22,23,24,25,26,27] have reported DTA curves including evidence for the presence stable cubic C3AH6. All report an endothermic peak due to dehydration somewhere in the range 300–330 °C. The products of hydration will also include alumina gel (AH3-gel) and gibbsite Al(OH)3 that decompose below 120 °C [18] and at a temperature of around 275 or 280 °C [19, 20], respectively.
It can be strongly argued that the importance of inorganic hydraulic binders is only exceeded by our lack of knowledge as to the factors influencing the synthesis, structures and properties of the calcium aluminates in CACs and calcium zirconium aluminate. One approach to developing this group of materials that can be taken is to examine the effect dopant cations, e.g. Sr2+, have on the properties and the hydraulic activity of Ca7ZrAl6O18. Some series of oxides Ca1−xSrxAl2O4 and Ca3−xSrxAl2O6 were found in the literature review [28,29,30]. Following this approach in this paper, the hydration behaviour of a series of oxides of the type Ca7−xSrxZrAl6O18 using isothermal calorimetry at room temperature is presented. The hydration products were controlled by means of X-ray diffraction analysis, thermal analysis (DTA–TG–EGA) and scanning electron microscopy (SEM/EDS) of the cement stone.
Experimental
Synthesis
The synthesis of a series of oxides of the type Ca7−xSrxZrAl6O18, where x = 0.3, 0.6, 1.0, and the reference material with x = 0 as undoped Ca7ZrAl6O18 was performed by a two-step firing procedure using two different sintering temperatures. In a first step, calcium carbonate (99.81% CaCO3, Chempur), strontium carbonate (99.00% SrCO3, Merck), α-Al2O3 (99.8% Al2O3, Across Organics) and zirconia (98.08% ZrO2, Merck) powders were mixed with appropriate molar ratio of CaO, SrO, Al2O3 and ZrO2 oxides, and then, the mixtures were homogenized for 2 h in a ball mill and pressed into cylinders having a diameter of 2 cm. All green pellets were calcined at 1300 °C for 10 h. In a second step, solid-state sintering of the pellets made from the calcined powder at 1420 °C for 15 h resulted in phases Sr2+-doped Ca7ZrAl6O18. An intermediate grinding and mixing stage in order to improve homogeneity was necessary. As a final step, the reference Ca7ZrAl6O18 and Sr2+-doped Ca7ZrAl6O18 sinters were ground to a fine powder using a mortar and pestle, and then, they were homogenized for 1 h in a ball mill. The samples were characterized by X-ray powder diffraction using X’Pert ProPANalytical X-ray diffractometer. The XRD patterns of powdered samples were collected by step scanning with step of 0.02° over the range 5°–90° at room temperature. The phases developed during sintering were compared and confirmed using search–match reference ICDD database. The pellets were also polished, coated with carbon and then analysed using Nova NanoSEM 200 scanning electron microscope.
Cementitious paste preparation and methods of investigation
The course of hydration of a series of oxides of the type Ca7−xSrxZrAl6O18, where x = 0.3, 0.6, 1.0, and the reference material with x = 0 as undoped Ca7ZrAl6O18 was investigated using isothermal microcalorimeter TAM Air (TA Instruments) by integrating the continuous heat flow signal during the 72-h hydration process. Calorimetric measurements of cementitious phases were taken using the in situ mixing treatments of powder with water with 1.0 of water/cement mass ratio (w/c) and temperature of 25 °C. All tests were performed by mixing the pastes inside the calorimeter to study the early hydration processes of cements. Two grams of dry powder was poured into a glass vial and water weighed into mounted syringes. Admix ampoule was introduced into the calorimeter prior mixing, thus equilibrating to isothermal environment. Mixing was done after equilibration inside the calorimeter to enable the quantitative access to the heat of the early hydration process. A second glass vial containing sand (providing the same heat capacity that the mass of binder paste) was used as an inert reference in the twin channel of calorimeter to ensure best stability of the baseline. Heat flow curves were normalized to the mass of total dry binder and reported in terms of power (mW g−1 dry binder) versus time (hours).
The hydration products and the thermal stability of hydrates were investigated by differential thermal analysis–thermogravimetric analysis–evolved gas analysis (DTA–TG–EGA), X-ray diffraction analysis and scanning electron microscopy–energy-dispersive spectroscopy (SEM–EDS) (mentioned above). For this purpose, the neat cement paste composed of Ca7−xSrxZrAl6O18, where x = 1.0, and water with w/c = 1.0 was homogenized by hand, in a glass baker. Sample in paste form was placed in sealed polyethylene bag and cured up to 21 days in a climatic chamber with the relative humidity maintained at 95% and temperature of 25 °C. As a next step, samples were dried by acetone quenching at specified intervals of time, i.e. 15 min, 0.5 h, 1 h, 24 h, 3 days, 7 days, 14 days and 21 days. Dry powders were then analysed by XRD and thermal analysis. A simultaneous DTA–TG–EGA method (NETZSCH STA 449 F5 Jupiter coupled to QMS 403 D Aëolos) at a heating rate of 10 °C min−1 under a flow of Ar (50 mL min−1) was applied. The containers for samples and reference were corundum crucibles. The reference corundum crucible was kept empty during all DTA–TG–EGA tests. The initial sample mass was 25 mg.
Characterization of microstructure evolution of cement paste by SEM was performed on the freshly broken surfaces of the hardened cement pastes after 24 h, 3 days and 11 days of curing duration. The samples were coated with carbon in order to remove any charge.
Results and discussion
Synthesis of the Sr2+-doped calcium zirconium aluminate cement
The samples with different concentrations of strontia sintered at 1420 °C for 15 h were ground, and the phases were identified by XRD analysis. The X-ray diffraction results present no diffraction peaks of SrO and other Sr-bearing phases admixtures, e.g. strontium aluminates. Moreover, the peaks of Ca7ZrAl6O18 shifted gradually with increasing the content of SrO, thus indicating that Sr2+ ions have been doped into Ca7ZrAl6O18 lattice to form the mixed oxides of the type Ca7−xSrxZrAl6O18 solid solutions, where x = 0.3, 0.6, 1.0. The XRD pattern for concentration of strontia x = 1.0 unhydrated composite will be presented later together with the powder XRD patterns for the Ca7−xSrxZrAl6O16 (x = 1.0)-based cement paste. Effect of Sr2+ ions doping on Ca7ZrAl6O18 lattice was confirmed by comparing the values of the observed peaks with their standard values as per JCPDS No. 98-018-2622 [31]. It is found that with the increase in the SrO content, a shift in the lines corresponds to a somewhat larger unit cell for the “pure” undoped Ca7ZrAl6O18 in comparison with Sr-doped Ca7ZrAl6O18 solid solutions since the substituting Sr2+ ion is larger than Ca2+ (1.13 vs. 0.99 Å) [28]. Figure 1a, b shows the results of the SEM and EDS observations of Ca7−xSrxZrAl6O16 (x = 1.0) sinter heat treated at 1420 °C for 15 h. Almost pure crystal phase of Sr2+-doped calcium zirconium aluminate with some amount of accessory (Ca,Sr)ZrO3 was found in the SEM microstructure. The EDS spectrum was taken on the region where strontium formed a solid solution within the matrix, which is shown as grey region in SEM image (Fig. 1b). The mass percentage of elements determined by the quantitative EDS microanalysis in SEM is 26.73 mass% Ca, 12.82 mass% Sr, 12.96 mass% Zr, 20.36 mass% Al and 27.14 mass% O. This results closely relate to the concentration of elements in the stoichiometric composition, designed as (Ca6Sr)ZrAl6O18.

a SEM image of the microstructure of as-synthesized Sr2+-doped Ca7ZrAl6O18 sample heat treated at 1420 °C for 15 h. b EDS spectrum and quantitative composition analysis of grey region in (a)
Hydration and crystalline hydration products of the Sr2+-doped calcium zirconium aluminate cement
The results of isothermal calorimetric experiment (Figs. 2, 3), X-ray diffraction analysis (Figs. 4, 5), simultaneous thermal analysis (Figs. 6, 7a, b) and scanning electron microscopy (Figs. 8–10) were used to study the evolution of phase composition during the process of hydration of Sr2+-doped calcium zirconium aluminate cement paste with w/c of 1.0 at room temperature. Figures 2 and 3 exhibit the influence of chemical and structural modification of Ca7ZrAl6O18 phase induced by strontium doping upon the calorimetric curves at constant value of water/cement ratio. Figure 2b demonstrates the rate of heat evolution for the blended cements over a 72-h period. As the Ca7ZrAl6O18 phase was doped with Sr2+ ions (Ca7−xSrxZrAl6O18; x = 0 (as undoped, reference), 0.3, 0.6, 1.0), the intensity of the peak heat rate decreased gradually with increasing strontium content (Fig. 2a). This figure presents not typical heat evolution rate with only one peak that corresponds to initial reactions which follow the mixing of the cement with water. There is also not any occurrence of induction period observed; hence, the hydration reaction starts immediately after mixing of these cementitious phases with water at room temperature. The first peak corresponds to initial reactions which follow the mixing of the cement with water. It records the heat of wetting, dissolution, hydration of Sr2+-doped calcium zirconium aluminate and the early formation of hydration products. There is no occurrence of the dormant period or the induction period in the heat flow curve (Fig. 2). After about 2 h, the hydration reaches a steady state and future hydration is diffusion controlled. Thus, the mass transport of water and dissolved ions through the hydration barrier controls hydration process. No other exothermic peaks were also registered during 72 h of experiment. Figure 3 shows an integration of the heat evolution over a period of 72 h. It is particularly easy to make the very clear conclusions that for given strontium-modified cements all hydration produces less heat as compared to pure Ca7ZrAl6O18. According to the initial structural model given by Fukuda et al. [5], the highly disordered crystal structure of Ca7ZrAl6O18 with the five types of positions of Ca atoms and four types of AlO4 tetrahedra was found. Ca atoms are located between [Al6O18] rings created by interconnected six AlO4 tetrahedra in this structure. The incorporation of Sr (instead of Ca) causes both chemical and structural modifications of Ca7ZrAl6O18, thus influencing its hydration kinetics.

Heat flow curves as a function of hydration time (a the first 2 h; b the period of 72 h) of the undoped reference Ca7ZrAl6O18 and the Ca7−xSrxZrAl6O16 (x = 0.3, 0.6 and 1.0) cement pastes prepared with 1.0 of w/c (water-to-cement) ratio and hydrated at room temperature, normalized to mass of binder paste (with in situ mixing condition)

Cumulative heat curves as a function of hydration time of the undoped reference Ca7ZrAl6O18 and the Ca7−xSrxZrAl6O16 (x = 0.3, 0.6 and 1.0) cement pastes prepared with 1.0 of w/c (water-to-cement) ratio and hydrated at room temperature for the period of 72 h

Powder XRD patterns for the Ca7−xSrxZrAl6O16 (x = 1.0)-based cement paste prepared with 1.0 of w/c ratio and hydrated at room temperature for the period of 0.5 h (b), 24 h (c), 7, 14 and 21 days (d–f). a Unhydrated Ca7−xSrxZrAl6O16 (x = 1.0) phase

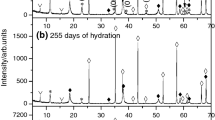
Powder XRD patterns for the Ca7−xSrxZrAl6O16 (x = 1.0)-based cement pastes collected at 2θ from 27° to 35°

Simultaneous TG (in grey)/DTA (in black) curves of cementitious binder samples measured in argon at flow rate 50 mL min−1 (heating rate 10 °C min−1, initial mass 25 mg)

Gas evolution curves for representative mass spectroscopic ion fragments of H2O (a) and CO2 (b) vapours during the thermal decomposition of the cementitious binder samples in flowing argon, measured in situ by online coupled TG/DTA–EGA–MS system (Ar flow 50 mL min−1, heating rate 10 °C min−1)

SEM images of the fracture surface of 24-h hydrated Sr2+-doped calcium zirconium aluminate cement paste (room temperature)
The course of heat evolution during hydration of cementitious phases described by means of calorimetric curves plotting the rate of heat evolution in mW g−1 can be pertained to changes in the phase composition of the hardened cement pastes controlled by means of X-ray diffraction (XRD).
The mineralogical composition of the acetone quenched hydrated cementitious pastes determined by XRD is presented in Figs. 4 and 5. The original sample is an unhydrated compound (Fig. 4a) that undergoes a phase transformation when it reacts with water at room temperature. The hydration process occurs in the Sr2+-doped calcium zirconium aluminate cement at various time intervals (0.5, 24 h; 7, 14 and 21 days). As indicated in Eq. (1), the overall reaction proceeds as chemical changes take place leading to the formation of two separate products (C,Sr)4AH19 and (C,Sr)2AH8 at the early stage of hydration. Since strontium ions can substitute the calcium in the hexagonal “pure” C–A–H and AFm phases [30, 32, 33], broadening of the diffraction peaks caused by structural disorder of layered metastable hydrates [34] was observed.
Hydration
The typical crystalline phases of first hydration products were hexagonal tetracalcium aluminate 19-hydrate ((C,Sr)4AH19) and dicalcium aluminate hydrate ((C,Sr)2AH8) that were observed after about 0.5 h of curing in cementitious paste as evidenced from the characteristic diffraction peaks centred at specific angles 2θ = 30.868° (d = 2.89448 Å) and 2θ = 8.2787° (d = 10.67150 Å). This is in good agreement with JCPDS cards No. 00-014-628 and No. 00-045-0564 for C4AH19 and C2AH8, respectively. Nevertheless, the X-ray diffraction patterns of (C,Sr)4AH19 and (C,Sr)2AH8 are very close making no further clear distinction between these two hydration products.
As time progresses, (C,Sr)4AH19 losses part of its interlayer water to form (C,Sr)4AH13, according to Eq. (2):
Partial dehydration
The formation of tetracalcium aluminium 13-hydrate (C,Sr)4AH13) between 24 h and 14 days in the Ca7−xSrxZrAl6O16 (x = 1.0)-based hydrated cement paste was identified by X-ray diffraction analysis, as shown in Fig. 4c–e. The diffraction patterns show the most detectable peak (the “100% peak”) of C4AH13 at about 2θ = 11.1638° (d = 7.91968 Å) that is in good agreement with reference data (JCPDS card No. 00-011-0203).
The interlayer exchange of OH− by CO3 2− proceeds via a stepwise manner such as shown in Eq. 3. This exchange is accompanied by a shift in the position of the diffraction maxima from 2θ = 11.164° to 2θ = 11.6293° (d = 7.60964 Å). The latest one belongs to the calcium carboaluminate hydrate C3A·CaCO3·11H2O (JCPDS cards No. 00-036-0377) which has been identified in 21-day cured cementitious paste.
Carbonation
In the conversion reaction, compounds (C,Sr)4AH19 (or (C,Sr)4AH13) and (C,Sr)2AH8 redissolved or interact to give (C,Sr)3AH6, hydrogarnet, along with additional AH3, which over time tends increasingly to approximate to gibbsite (Eqs. 4a–c).
Transformation
The powder X-ray patterns of 24-h–14-day cured cementitious paste and 21-day cured cementitious paste indicate that at least two different kinds of components of hydrogarnet were present in these samples (Fig. 4c–f). In essence, the diffraction patterns of a sample of Ca7−xSrxZrAl6O16 (x = 1.0), treated with water within the curing time range between 24 h and 14 days (Fig. 4c–e) and hydrated up to 21 days (Fig. 4f), show the presence of two series of similar lines that are located very close together, indicating that two hydration products having a similar crystal structure are formed. Going into details, two isostructural hydrates were formed like Sr-rich and Ca-rich cubic phase (C,Sr)3AH6. The position of characteristic line of Sr-rich cubic phase (C,Sr)3AH6 is very close to the location of Ca-rich one, but it was found at lower values of 2theta. All diffraction lines in powder patterns belonging generally to cubic phase, as compared with standard X-ray diffraction pattern of C3AH6 (JCPDS card No. 01-071-0735), are shifted to lower values of 2theta. There are two phenomena in the presented results. Firstly, the three most intense peaks at 16.7921°, 38.1904° and 31.0501° are due to the Ca-rich cubic phase (C,Sr)3AH6 in the XRD pattern of Ca7−xSrxZrAl6O16 (x = 1.0) cement paste prepared with 1.0 of w/c ratio and hydrated at room temperature for the period of 21 days (Fig. 4f). Secondly, Fig. 4c–e clearly emphasizes the formation of a second hydrogarnet phase, Sr-rich cubic (C,Sr)3AH6, as evidenced from the diffraction peaks positioned at 16.5731°, 37.9245° and 30.8052° of 2θ for 14-day cured sample, given as an example. The significant 2θ peak shifts to lower angles when Sr2+ was used, with respect to peaks from “pure” C3AH6, and formation of both Sr-rich (C,Sr)3AH6 and Ca-rich (C,Sr)3AH6-type solid solutions could be attributed to induced crystal-field effects due to the different ionic size of Ca2+ (0.99 Å) compared with Sr2+ (1.13 Å) [28]. Hence, Sr-rich (C,Sr)3AH6 was stable within a reaction period from 24 h to 14 days (Fig. 4c–e). The consecutive formation of two separate solid solution members, supported by Ref. [28], one close to the strontium (Sr-rich (C,Sr)3AH6) and the other to the calcium (Ca-rich (C,Sr)3AH6) end, leads to the conclusion that the strontium reacted first, by reason of its greater solubility, leaving a solution richer in lime to form later. Shifting to the left from the standard XRD peaks of C3AH6 due to replacing Ca2+ with Sr2+ is evident from both Fig. 5c–f and Table 1. Moreover, residues of unhydrated (Ca,Sr)7ZrAl6O18 were observed up to 14 days of curing, and this phase continues to hydrate slowly over the next 7 days (Fig. 5e–f).
Thermal stability of hydration products of the Sr2+-doped calcium zirconium aluminate cement paste
The simultaneous TG/DTA measurements show several decomposition stages of the Sr2+-doped calcium zirconium aluminate cement paste after curing time period of 15 min to 21 days (Fig. 6). The progress of cement hydration consumes water and produces hydrates; thus, the highest loss of mass due to the evaporation of the water chemically bonded to C–Sr–A–H phases was achieved after 21 days of moist curing. The total mass loss due to decomposition of the cement paste between about 25 and 1000 °C is 9.89, 10.23, 12.98, 28.71, 32.05, 35.53, 35.59 and 38.00% of the initial mass, corresponding to the length of curing time according to thermogravimetric analysis results displayed as TG curves (in grey) for samples cured as long as 15 min, 0.5 h, 1 h, 24 h, 3 days, 7 days, 14 days and 21 days, respectively. These curves can be compared to the in situ analysis of the evolved gaseous species by the online coupled EGA–MS system. Main evolution curves of H2O (m/z = 18) and CO2 (m/z = 44) are also shown as ion current versus temperature in Fig. 7a, b. Evolution of H2O shows parallel run to the DTA curves having local maximums at the same temperature values. Nevertheless, the endothermic dehydration peaks are more sensitively detected by the MS than DTA curves and the characteristic peaks of evolution of H2O are clearly visible in all the samples. In the temperature range of 25–1000 °C, the peaks attributed to H2O are apparently strengthened, thus confirming the water consumed in the formation of the hydration products with hydration time. The signals for H2O change from one peak at 259 °C to strong peaks between 90 and 310 °C. The EGA peak attributed to H2O at 259, 258 or 266 °C was due to the decomposition reaction of the crystalline (C,Sr)4AH19, which was formed preferentially during the first 15 min and exists up to ca. 1 h of hydration (Fig. 7a). Next, fraction of bound water of (C,Sr)4AH19 was lost, and within a time range between 24 h and 7 days, the (C,Sr)4AH19 lost part of its interlayer water to form (C,Sr)4AH13. At the temperature of 273 or 274 °C, the peaks attributed to H2O due to the decomposition reaction of (C,Sr)4AH13 were observed in the mass spectroscopic gas-evolution curves of the cementitious pastes hydrated between 24 h and 7 days (Fig. 7a). Starting from 14 days of hydration, this peak was not observed clearly in the m/z 18 curve that proves the partial disappearance of (C,Sr)4AH13 phase including its carbonation and formation of the crystalline \(\left( {\text{C,Sr}} \right)_{4} {\text{A}}\bar{C}{\text{H}}_{11}\) phase within a time range between 14 and 21 days. The peak of tetracalcium aluminium carbonate hydrate coincides with the peak (C,Sr)2AH8 at about 178 °C. The EGA peak attributed to H2O evolution at 158, 166, 173, 179 or 178 °C was due to the decomposition reaction of the crystalline (C,Sr)2AH8, which was detected within a time range between 0.5 h and 21 days (Fig. 7a). The gradual shift of these peaks to higher temperature as curing time progresses may be caused by an increase in the degree of crystallinity this hydrate.
The water release from Sr-rich (C,Sr)3AH6 and Ca-rich (C,Sr)3AH6 begins as soon as the conversion of metastable hydrates to the stable higher-density phases starts after 24 h of hydration and it is visible as EGA peak at ca. 300 °C in the evolution curves of H2O of the Sr2+-doped calcium zirconium aluminate cement paste hydrated within a time range between 24 h and 21 days (Fig. 7a). Despite the fact that the cubic phases Sr-rich (C,Sr)3AH6 and Ca-rich (C,Sr)3AH6 were clearly distinguished from the XRD patterns of both 14-day and 21-day hydrated cement pastes as shown in Figs. 4, 5, there is no clear difference in the EGA peaks at 309 and 308 °C (Fig. 7a), respectively.
According to the DTA, TG and EGA curves, the loss of mass due to the decomposition of AH3 gel takes place below 120 °C with the broad endothermic effect in all blended cement pastes but is clearly visible as a strong effect in samples cured for at least 24 h. The decomposition step due to the dehydration of crystalline gibbsite Al(OH)3 is observed in Fig. 7a as the EGA peaks at 273 °C that precede the peak of Ca-rich (C,Sr)3AH6 under long-term curing age up to 21 days. The evolution of CO2 gas from the carbonated Sr2+-doped calcium zirconium aluminate cement consisted of different stages with broad EGA peaks (Fig. 7b).
Microstructural investigations of the Sr2+-doped calcium zirconium aluminate cement paste
The microstructure of hydration products of the Sr2+-doped calcium zirconium aluminate binder paste was examined by SEM/EDS (Figs. 8–10a). The SEM micrographs in Fig. 8 show that the flake-like crystals of C–Sr–A–H and amorphous gels are the major hexagonal hydration products and they were formed at age of 24 h at room temperature. As hydration proceeds, the unhydrated grain core is continuously consumed to form hydration products and the hydrated product of the Sr2+-doped calcium zirconium aluminate compound in a grain of cement adheres firmly to the unhydrated core of the grains of cement as shown in Figs. 9 and 10a. The characteristics of the hydrates were examined by EDS analysis after the SEM examination, as illustrated in Fig. 10b and Table 2. These results show that the hydrated matrix contains calcium, strontium and aluminium, which can be related to the calcium strontium aluminium hydrates (C–Sr–A–H). There was also a clear segregation of zirconium within the reaction zone generated from unhydrated grain core of (Ca,Sr)7ZrAl6O18 to the hydrated matrix C–Sr–A–H (points 1–2–3).

SEM images of the fracture surface of 3-day hydrated Sr2+-doped calcium zirconium aluminate cement paste (room temperature)

SEM images of the fracture surface of 11-day hydrated Sr2+-doped calcium zirconium aluminate cement paste (room temperature). Spot 1–3 EDS analysis. a EDS spectrum of sample in the micro area 3
Conclusions
-
1.
Since no diffraction peaks of SrO and other Sr-bearing phases, e.g. strontium aluminates, can be detected in samples and the peaks of Ca7ZrAl6O18 shift gradually with the composition, it indicates that SrO has been doped into Ca7ZrAl6O18 lattice to form uniform solid solution.
-
2.
Intensity of the first peak of heat evolution is gradually reduced as the x value in the Ca7−xSrxZrAl6O18 (x = 0.3, 0.6, 1.0) formula increases.
-
3.
The hydration heat first peak of the mixed oxides of the type 6CaO·SrO·3Al2O3·ZrO2 is generated simultaneously with both the wetting of cement grains and the formation of Sr-doped hexagonal C4AH13–19 and C2AH8 hydration products.
-
4.
Including strontium ions in the solid solutions of unstable hexagonal hydrates could effectively inhibit the conversion from both C4AH13–19 and C2AH8 to stable C3AH6 and AH3 products.
-
5.
The new phase C3A·CaCO3·11H2O (\({\text{C}}_{4} {\text{A}}\bar{C}{\text{H}}_{11}\)) was found in this system due to the activity of carbon dioxide; the formation of C4AH13–19 and C2AH8 is prohibited and the generation of stable cubic phase is delayed in the early hydration process.
-
6.
Two hydrogarnet phases, one Sr-rich (C,Sr)3AH6 and the other Ca-rich (C,Sr)3AH6, were formed due to the inevitable and irreversible conversion of metastable hydrates.
References
Pacewska B, Nowacka M. Studies of conversion progress of calcium aluminate cement hydrates by thermal analysis method. J Therm Anal Calorim. 2014;117(2):653–60.
Boris R, Antonovič V, Keriene J, Stonys R. The effect of carbon fiber additive on early hydration of calcium aluminate cement. J Therm Anal Calorim. 2016;125:1061–70.
Berezhnoi AS, Kordyuk RA. Melting diagram of the system CaO–Al2O3–ZrO2. Dopov Akad Nauk URSR. 1963;10:1344–7.
Berezhnoi AS, Tarnopol’skaya RA. Calcium aluminozirconate—a new hydraulic binder. Neorg Mater Akad Nauk. 1968;4:2151–4.
Fukuda K, Iwata T, Nishiyuki K. Crystal Structure, structural disorder, and hydration behavior of calcium zirconium aluminate, Ca7ZrAl6O18. Chem Mater. 2007;19(15):3726–31.
Madej D, Szczerba J, Nocuń-Wczelik W, Gajerski R. Hydration of Ca7ZrAl6O18 phase. Ceram Int. 2012;38(5):3821–7.
Madej D, Szczerba J. Study of the hydration of calcium zirconium aluminate (Ca7ZrAl6O18) blended with reactive alumina by calorimetry, thermogravimetry and other methods. J Therm Anal Calorim. 2015;121:579–88.
Szczerba J, Madej D, Śnieżek E, Prorok R. The application of DTA and TG methods to investigate of the non-crystalline hydration products of CaAl2O4 and Ca7ZrAl6O18 compounds. Thermochim Acta. 2013;567(10):40–5.
Madej D, Szczerba J, Nocuń-Wczelik W, Gajerski R, Hodur K. Studies on thermal dehydration of the hydrated Ca7ZrAl6O18 at different water-solid ratios cured at 60°C. Thermochim Acta. 2013;569:55–60.
Madej D, Szczerba J, Dul K. Phase transformation during the decomposition of hydrated calcium zirconium aluminate (Ca7ZrAl6O18) paste subjected to various dehydration temperatures. Thermochim Acta. 2014;597(10):27–34.
Madej D, Szczerba J. Preparation of Al2O3–CaAl12O19–ZrO2 composite ceramic material by the hydration and sintering of Ca7ZrAl6O18-reactive alumina mixture. Ceram-Silik. 2016;60(1):27–33.
Szczerba J, Madej D, Dul K, Bobowska P. Ca7ZrAl6O18 acting as a hydraulic and ceramic bonding in the MgO–CaZrO3 dense refractory composite. Ceram Int. 2014;40(5):7315–20.
Taylor HFW. Cement chemistry. 2nd ed. London: Thomas Telford; 1997.
Hewlett PC. Lea’s chemistry of cement and concrete. 4th ed. London: Elsevier Science and Technology Books; 2004.
Barnes P, Bensted J. Structure and Performance of Cements. 2nd ed. London: CRC Press; 2001.
Флeйшep ГЮ, Toкapчyк BB, Cвiдepcький BA. The effect of nitrogen-containing organic admixtures on the chemical processes of cement hardening. East Eur J Enterp Technol. 2016;1(6(79)):46–54.
Hill RL. The study of hydration of fly ash in the presence of calcium nitrate and calcium formate. Doctoral dissertation. Texas; 1994 (unpublished research work).
Bushnell-Watson SM, Sharp JH. The detection of the carboaluminate phase in hydrated high alumina cements by differential thermal analysis. Thermochim Acta. 1985;93:613–6.
George CM. Industrial aluminous cements. In: Barnes P, editor. Structure and Performance of Cements. London: Appl Sci; 1983. p. 415–70.
Midgley HG. Measurement of high-alumina cement-calcium carbonate reactions using DTA. Clay Miner. 1984;19:857–64.
Das SK, Mitra A, Dad Padar PK. Thermal analysis of hydrated calcium aluminates. J Therm Anal. 1996;47:765–74.
Fryda H, Scrivener KL, Chanvillard G. Relevance of laboratory tests to field applications of calcium aluminate cement concretes. In: Mangabhai RJ, Glasser FP, editors. Calcium aluminate cements. London: IOM Communications; 2001. p. 227–46.
Ewais EMM, Khalil NM, Amin MS, Ahmed YMZ, Barakat MA. Utilization of aluminum sludge and aluminum slag (dross) for the manufacture of calcium aluminate cement. Ceram Int. 2009;35:3381–8.
Abo-El-Enein SA, Heikal M, Amin MS, Negm HH. Reactivity of dealuminated kaolin and burnt kaolin using cement kiln dust or hydrated lime as activators. Constr Build Mater. 2013;47:1451–60.
Hashem FS, Amin MS, El-Gamal SMA. Improvement of acid resistance of Portland cement pastes using rice husk ash and cement kiln dust as additives. J Therm Anal Calorim. 2013;111:1391–8.
Amin MS, El-Gamal SMA, Hashem FS. Fire resistance and mechanical properties of carbon nanotubes—clay bricks wastes (Homra) composites cement. Constr Build Mater. 2015;98:237–49.
Abo-El-Enein SA, Hashem FS, Amin MS, Sayed DM. Physicochemical characteristics of cementitious building materials derived from industrial solid wastes. Constr Build Mater. 2016;126:983–90.
Prodjosantoso AK, Kennedy BJ, Hunter BA. Phase separation induced by hydration of the mixed Ca/Sr aluminates Ca3−xSrxAl2O6. Cement Concrete Res. 2002;32:647–55.
Prodjosantoso AK, Kennedy BJ. Synthesis and evolution of the crystalline phases in Ca1−xSrxAl2O4. J Solid State Chem. 2002;168:229–36.
Pöllmann H, Kaden R. Mono- (strontium-, calcium-) aluminate based cements. In: Fentiman CH, Mangabhai RJ, Scrivener KL, editors. Calcium aluminates proceedings of the international conference. Garston: IHS BRE Press; 2014. p. 99–108.
ICDD and ICSD PDF-2 database products.
Atkins M, Glasser FP. Application of Portland cement-based materials to radioactive waste immobilization. Waste Manage. 1992;12:105–31.
Evans NDM. Binding mechanisms of radionuclides to cement. Cement Concrete Res. 2008;38:543–53.
Britto S, Joseph S, Kamath PV. Distinguishing crystallite size effects from those of structural disorder on the powder X-ray diffraction patterns of layered materials. J Chem Sci. 2010;122(5):751–6.
Acknowledgements
The research was performed at Faculty of Materials Science and Ceramics of AGH and was supported by the Project No. 11.11.160.617.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Madej, D. Hydration, carbonation and thermal stability of hydrates in Ca7−xSrxZrAl6O18 cement. J Therm Anal Calorim 131, 2411–2420 (2018). https://doi.org/10.1007/s10973-017-6726-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6726-1