
Abstract
In this paper, ionic liquid treatment was applied to produce nanometric cellulose particles of two polymorphic forms. A complex characterization of nanofillers including wide-angle X-ray scattering, Fourier transform infrared spectroscopy, and particle size determination was performed. The evaluated ionic liquid treatment was effective in terms of nanocrystalline cellulose production, leaving chemical and supermolecular structure of the materials intact. However, nanocrystalline cellulose II was found to be more prone to ionic liquid hydrolysis leading to formation larger amount of small particles. Each nanocrystalline cellulose was subsequently mixed with a solution of chitosan, so that composite films containing 1, 3, and 5% mass/mass of nanometric filler were obtained. Reference samples of chitosan and chitosan with micrometric celluloses were also solvent casted. Thermal, mechanical, and morphological properties of films were tested and correlated with properties of filler used. The results of both, tensile tests and thermogravimetric analysis showed a significant discrepancy between composites filled with nanocrystalline cellulose I and nanocrystalline cellulose II.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rapidly developing technologies for manufacturing of functional and advanced material put a growing pressure on production of materials with specifically defined and unique properties. Preparation of biomaterials based on polymers of natural origin, which include cellulose, especially nanometric cellulose, is of particular importance. Cellulose is the most abundant natural polymer that is of high interest among researchers. The main challenge regarding nanometric cellulose is finding a way of its production that would result in a product showing high surface area, high aspect ratio, low density, and great mechanical properties. For example, nanocrystals of cellulose or whiskers are characterized with Young modulus in the range from 130 to 145 GPa [1]. Great interest in such materials is caused by their interesting and unique properties. Nanoparticles of cellulose undergo degradation faster than macroscopic cellulose, whereas other important and widely used nanoparticles such as fullerenes and nanotubes do not undergo biodegradation at all [2]. When compared to micrometric cellulose higher surface area and presence of nanopores in nanometric cellulose causes, the increase in interactions with other substances resulting in linkage with various nanoparticles [3]. Advantages of nanometric cellulose are not only connected with its physical and chemical properties or its susceptibility to degradation but also with its high biocompatibility and availability, renewability of raw material, and sustainable growth. It is used in aerogels, adhesive materials or as an additive for shape memory segmented polyurethanes modifying its thermal properties [4, 5]. Due to its ability to orientate in magnetic field, cellulose is also used in liquid crystal systems [6]. Currently, nanometric cellulose is most commonly produced by mechanical treatment or traditional acidic hydrolysis that in first stage is responsible for the disintegration of glycoside bonds in amorphous regions. Properties of obtained biomaterials depend on morphology and structure of nanometric cellulose what is closely connected with hydrolysis process of cellulosic materials. Controlling the size and shape of nanometric cellulose is crucial for production of nanometric cellulose characterized with unique and defined properties and therefore for its application as functional filler for polymers. That is possible only if controlled dissolution or controlled enzymatic etching of cellulose takes place. Acidic hydrolysis is mainly realized with sulfuric acid, hydrochloric acid, or hydrogen bromide. This method has several drawbacks, including difficulties in controlling the progress of the reaction [7, 8] or quite intensive formation of sulfate ester groups at the surface of nanometric cellulose that in turn influences interactions at the nanometric cellulose/polymer matrix interface and decreases its thermal stability [9]. Not without significance is the necessity to use concentrated, toxic reagents and the subsequent separation of nanoparticles from such solutions. Hence, ionic liquids are suggested as an alternative hydrolyzing agent.
Room temperature ionic liquids are a unique group of organic salts consisting of organic cation and disproportionate organic or nonorganic anion, with melting temperature below 100 °C [10]. They show an ability to dissolve both organic and nonorganic matter, and thus they are often labeled as new generation or “green solvents” [11]. Despite the fact that the mechanism of cellulose dissolution with ionic liquid remains disputable, some ionic liquids, favorably imidazole-based, are used to produce nanometric cellulose. Moreover, it was proved that aqueous ionic liquid is as well or even more effective in terms of nanometric cellulose production than dehydrated ones [12, 13]. Time and temperature of ionic liquid treatment were also found to be of critical importance [14]. Even though ionic liquids offer a great opportunity for production of nanocrystalline cellulose (higher degree of homogeneity, less aggressive reaction medium, medium recovery, etc.), still only few papers regarding application of, e.g., 1-butyl-3-methylimidazolium chloride or 1-butyl-3-methylimidazolium hydrogen sulfate for that purpose were published [14–16]. The reason is that currently the main aim of most of the research is to find new ionic liquids that enhance the effectiveness of dissolution of cellulose coming from lignocellulose materials [17–23] rather than the preparation of nanometric cellulose with ionic liquids. Ionic liquids are also relatively expensive, but they are recyclable, what can make the production of nanocrystalline cellulose more economically feasible and less environmentally adverse [24].
When aiming to produce nanometric cellulose, it should be also taken into consideration that starting material, cellulose, may exist in two polymorphic forms. This forms called cellulose I and cellulose II have a different arrangement of polysaccharide chains and different sizes of elementary cell [1]. It is also important that cellulose II is characterized with much lower degree of crystallinity than cellulose I. There are some papers in which the influence of cellulose type on nucleation and crystallization process of semicrystalline polymer matrices was reported as significant [25, 26]. That diversity of crystalline structures of both celluloses may also have a great influence on hydrolysis of cellulose and effectiveness of nanometric cellulose production. Likewise, divergent structure of hydrogen bonds in cellulose I and cellulose II is responsible for differences in thermal stability of these two crystalline forms [27]. It was already shown that in case of the acid hydrolysis of cellulose its polymorphic form has an influence on the particles sizes of produced nanometric celluloses. The higher yield of nanocrystalline cellulose II was ascribed to different crystallographic structures of starting celluloses and lower crystallinity of cellulose II, which was more susceptible to acid hydrolysis than cellulose I [28]. Still, the influence of polymorphic forms of cellulose on formation of cellulose nanocrystals using ionic liquids has not been reported yet.
Attractive mechanical properties of nanometric cellulose make it ideal filler for polymers. What is more, incorporation of nanometric cellulose particles in natural polymer is an attempt to mimic the nature. Combining nanometric cellulose with chitosan seems to be particularly interesting. Chitosan is a compound of marine origin, obtained by partial deacetylation of chitin. It consists of N-acetyl glucosamine and d-glucosamine units which are a source of amine and hydroxyl groups. Such chitosan/nanometric cellulose composite merges the properties of chitosan (biodegradability, antibacterial properties, transparency, antimicrobial activity) and nanometric cellulose (high surface area, very good barrier, and mechanical properties) [29–32] resulting in obtainment of composite materials that can be successfully applied in packaging industry (film for food, paper coatings), in chemical industry (catalysts, adsorbents), and in biomedicine (carrier of active substances, filaments) [33–36]. Despite of these valuable advantages, there are also compelling problems hindering wider application of such chitosan-based composites. Chitosan exhibits a high sensitivity to numerous types of degradation, including thermodegradation. Thermal analysis showed that this biopolymer cannot withstand temperature higher than 200–220 °C [37]. This poor thermal stability of chitosan-based systems often limits its wider application, but it can be overcome by addition of compounds that act as stabilizing agent. There are some papers which report an improvement of thermal stability of the chitosan with hydroxyapatite, nanoclay [38], or calcium carbonate [39]. Nanofibrillated cellulose produced using enzymes was also successfully used to enhance the thermal stability of the chitosan films [40].
In the light of the above considerations, it is interesting to study whether the incorporation of two different polymorphic forms of nanocrystalline cellulose produced only with ionic liquid can improve thermal and mechanical properties of chitosan-based composites. Although the literature contains some examples of the use of nanocrystalline cellulose as a reinforcing agent for chitosan composites, there are no reports on influence of polymorphic variety of cellulose produced by means of ionic liquid hydrolysis on properties of chitosan-based composites at the time of writing this article. The knowledge of this subject seems to be essential for designing new biomaterials with enhanced properties and wider potential applications. For that reason, the main purpose of this research was to evaluate the influence of crystallographic form of nanocrystalline cellulose produced with ionic liquid on thermal and mechanical properties of chitosan-based composites.
Experimental
Raw materials
Cellulose Avicel PH-101 with average particle size of 50 µm and high molecular weight chitosan from crab shells (degree of deacetylation 75–85%) was purchased from Sigma-Aldrich. Pure sodium hydroxide (Chempur) was used for preparation of 16% solution and subsequently used as mercerizing agent. Acetic acid 80% purchased from POCH S.A. was diluted to 2% (v/v) solution and used for composites formation. Ionic liquid 1-butyl-3-methylimidazolium hydrogen sulfate (bmimHSO4) with water content ≤1.0% was also purchased from POCH S.A.
Cellulose pulp mercerization
Cellulose (C I) pulp was treated with 16% NaOH at room temperature. After 5 min of continuous stirring, an excess of water was added to stop the mercerization process. The suspension was centrifuged at 10.000 rpm for 10 min. Finally, the produced cellulose II (C II) was washed with the excess of distilled water to remove NaOH and then dried in the air at 70 °C for 6 h.
Nanocrystalline cellulose I preparation
Nanocrystalline cellulose I (CNC I) was obtained through controlled ionic liquid hydrolysis of cellulose. Cellulose I was immersed in tenfold weight excess of bmimHSO4 for 36 h. After that time, water was added so that ionic liquid–water ratio was 80:20 mass/mass. As the temperature of 90 °C was reached, the reaction was continued for next 12 h under constant stirring at 400 rpm. Subsequently, the hydrolysis was stopped by adding water. The suspension was neutralized with significant amounts of water until pH ≈ 7 was reached, then sedimented, centrifuged, and filtered. Produced CNC I was dried at 70 °C for 6 h.
Nanocrystalline II preparation
Nanocrystalline cellulose II (CNC II) was prepared in the same manner as CNC I with that difference that C II was used as a starting material.
Composites preparation
Chitosan/nanocrystalline cellulose composites were produced by solvent casting method. Firstly, chitosan was dissolved in 2% (v/v) CH3COOH. Secondly, nanocrystalline celluloses at different concentrations were added to chitosan so that mixtures containing 1, 3, and 5% mass/mass of CNC (in relation to dry mass of chitosan) were obtained. Also, as comparison, composites with 5% of cellulose I and cellulose II were prepared. All mixtures were homogenized by ultrasonication for 20 min, applied on Petri dishes, and dried for 12 h at 35 °C. The samples were named according to the following convention: CHT stands for chitosan, number stands for percentage addition of filler, and C I, C II, CNC I, or CNC II stands for filler type. For example, name CHT/5 CNC II means chitosan with 5% loading of nanocrystalline cellulose II.
Fourier transform infrared spectroscopy (FTIR)
FTIR spectra confirming the composition of samples were recorded on an ATI Mattson Infinity Series FTIR spectrometer equipped with a deuterated triglycine sulfate detector in range from 500 to 4000 cm−1.
Wide-angle X-ray scattering (WAXS)
Structures of nanocrystalline celluloses were analyzed by means of wide-angle X-ray scattering (WAXS) using CuKα radiation at 30 kV and 25 mA anode excitation. The X-ray diffraction patterns were recorded for the angle range of from 5° to 30° in the step of 0.04°/3 s.
Particle size distribution
Mastersizer 2000 (Malvern Instruments Ltd.) and Zetasizer Nano ZS (Malvern Instruments Ltd.) employing the laser diffraction technique in the range of 0.2–2000 μm and 0.6–6000 nm, respectively, were applied to determine particle size and the dispersive properties of (nanometric) celluloses.
Thermogravimetric analysis (TG)
A thermogravimetric analyzer (Jupiter STA 449F3, Netzsch) was used to investigate the influence of filler type on thermal stability of the composites. Measurements were taken in the atmosphere of nitrogen (flow rate 20 cm3 min−1) at a heating rate of 10 K min−1 over a temperature range of 30–1100 °C, with an initial sample weight of approximately 5 mg.
Scanning electron microscopy (SEM)
The morphology of chitosan-based samples was studied using scanning electron microscopy (Hitachi TM3030Plus SEM) at acceleration voltage of 15 kV, using backscatter and secondary electron detectors. The specimens were not coated.
Tensile properties
Tensile properties of produced composite films were defined using Zwick and Roell Allround-Line Z020 TEW testing machine. Samples of 10 mm width and thickness ca. 0.1 mm were tested with speed 5 mm/min and initial force 0.2 N in accordance with standard ISO 527-3. The arithmetic mean of seven replicate determinations was taken into consideration in each case.
Results and discussion
Supermolecular and chemical structure of fillers
The effect of mercerization on supermolecular structures of produced nanocrystalline celluloses was examined by means of WAXS technique (Fig. 1). Also, starting materials—C I and C II—are shown in Fig. 1.

WAXS patterns for celluloses and nanocrystalline celluloses
The diffraction patterns of CNC I showed peaks at approximately 2Θ = 15°, 17°, 22.7° which are characteristic for structure of cellulose I. The crystalline structure of the mercerized cellulose was assigned to cellulose II because peaks at 2Θ = 12°, 20°, 22° were recorded. Since the WAXS pattern for CNC II exhibits not peaks coming from native cellulose, it is apparent that alkali treatment was successful and 100% of C I was transformed into C II. It is consistent with the literature that reports that NaOH concentrations higher than 10% are responsible for mercerization of cellulose I and therefore its transformation into cellulose II [41]. It is known that mercerization process causes an intermolecular rearrangement between the individual chains of cellulose and in result its crystallinity degree decreases. On the other hand, intensive ionic liquid pretreatment reduces the degree of cellulose crystallinity [42]. Both of these statements are confirmed with findings of WAXS analysis.
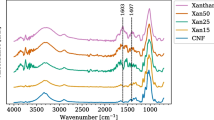
FTIR spectra of nanocrystalline cellulose fillers (Fig. 2) were recorded in order to confirm their chemical structures. Table 1 presents assignments of signals to the vibrations.

FTIR spectra of nanocrystalline celluloses
FTIR analysis showed main characteristic peaks of cellulose in both samples. At the same time, no traces of imidazole ring coming from ionic liquid were present proving that produced celluloses were sufficiently washed and that ionic liquid did not change the chemical structure of materials. Moreover, the lack of bands at 1731 cm−1 and approximately 1513 and 1239 cm−1 implies that no lignin, pectin, or hemicelluloses were present in the samples.
Dispersive and morphological properties of fillers
So far, in this paper, starting materials were called celluloses, while produced cellulosic materials were named nanocrystalline celluloses. Now, we would like to present the results proving the correctness of the terms mentioned above. Hence, particle size distributions of celluloses are presented in Fig. 3, and the Zetasizer Nano ZS results describing particles sizes of the obtained fillers are shown in Fig. 4.

Particle size distribution for a) C I and b) C II

Particle size distribution of CNC I and CNC II
Particle size distributions of celluloses (Fig. 3) show that mercerization process caused the decrease in particle sizes from ~48 µm for C I up to ~40 µm for C II. Despite this fact, produced particles were still in micrometric range, and thus further treatment leading to the production of nanometric cellulose was necessary.
Ionic liquid treatment of micrometric celluloses caused the formation of both nanometric particles and micrometric agglomerates (Fig. 4).
Sample of CNC I was almost equally divided into two fractions, while sample of CNC II was less homogenous, but in general four diameter ranges can be distinguished. When considering diameters ≤122 nm, it can be noticed that 42% of CNC I particles were in this size range, while for CNC II it was 28.2%. In the second range, from 142 up to 220 nm, particles of CNC II dominate (almost 22% of the samples, twice as much as for CNC I). If we move to the third range, 255–1900 nm, here no particles of CNC I were observed, but over 23% volume of the CNC II sample was this size. In the last, fourth range (≤2300 nm), only 26.1% of CNC II and 47.2% of CNC I, was found. Accordingly, it can be concluded that sample of CNC I consisted of slightly larger amount of particles up to 220 nm, but it was also characterized with large volume of micrometric fraction. In the study on acidic hydrolysis of cellulose I and cellulose II [28], it was found that even though fraction of CNC II nanometric particles was higher than of CNC I, the micrometric particles of CNC I were significantly bigger than those of CNC II (max. 3580 and 1990 nm for CNC I and CNC II, respectively). Similarly, ionic liquid treatment led to formation of CNC I particles with diameter of 4800 nm, whereas the biggest particles of CNC II had only 3090 nm. In case of both, ionic liquid and acidic hydrolysis, it can be stated that native cellulose is more prone to form agglomerates. It seems that the main resemblance in the ionic liquid and acid treatment is the formation of large micrometric fraction of CNC I.
The key to understanding the differences in sizes of CNC I and CNC II derived from ionic liquid treatment is the mechanism of cellulose dissolution (formation of nanometric cellulose is a controlled process of dissolution) with reference to crystalline structures of starting celluloses. The dissolution mechanism of cellulose in ionic liquids was long believed to be based on hydrogen bonds interactions between the anion of the ionic liquid and the hydroxyl groups of cellulose [43]. Lately, it was shown that also cation can play an important role in the dissolution process. This mechanism involves the synergistic interaction of cation and anion of ionic liquid and can be explained as follows: Firstly, anions bind to the hydroxyl groups on the surface of the cellulose, causing the weakening of the hydrogen bonds between cellulose chains. Secondly, the separation of cellulose chains is promoted by the intercalation of cations that are attracted to anions because of their opposite charge. It is also known, that the intercalation of the cation into cellulose network is easier if the molecule of anion is big (HSO4 − > Cl−) [44]. If we take a closer look at the processes taking place during mercerization, it turns out that during this treatment conformation of the cellulose chains changes. In result, loosening of the structure along with decrystallization occurs. Consequently, it can be assumed that, due to the loosening the structure and development of the amorphous regions which absorb chemicals easier than highly compacted crystalline regions [45], mercerization facilitates the intercalation of cation coming from ionic liquid. That explains why sample of CNC II consisted of smaller amount of micrometric particles than CNC I. Yet, the aspect of hydrogen bonds present in cellulose II cannot be omitted. The antiparallel chain model of C II is responsible for formation not only interchain, but also of interplane hydrogen bonds, making its hydrogen bonding system more complex compared to that of C I [46]. Still, it is suspected that hydrogen bonds between ionic liquid and C II are much stronger than interplane hydrogen bonds of C II, enabling successful separation of cellulose chains.
Thermal stability of composites
The TG curves of chitosan and its composites with micro- and nanometric celluloses are given in Fig. 5, while the corresponding tabulated values obtained from these measurements are provided in Table 2.

TG curves for chitosan and its composites
All the TG curves are smooth, with only two mass loss steps which indicates that the thermal degradation of chitosan and chitosan-based composites in nitrogen atmosphere is simple and, apart from water loss, is a one-step reaction. The first loss around 100 °C is associated with the evaporation of a residual acetic acid as well as water that is physically absorbed and strongly hydrogen bonded to chitosan and cellulosic fillers. As shown in Table 2, 5% mass loss for pristine chitosan was noted at significantly higher temperature than for its composites. It suggests that the evaluated composites contained more bonded water. The reason for that was the presence of cellulosic filler and cellulose is a hydrophilic material which is well known for being able to absorb high amounts of water. In general, it is believed that, because of the contrasting sorption mechanisms (bulk for C II and surface for C I), less crystalline C II can adsorb more water than C I [47, 48]. This relation can be seen for samples of CHT/5 C I and CHT/5 CII, but in our case it is no longer noticeable for composites with nanometric filler. The second stage (170–400 °C) of thermal decomposition is assigned to the degradation of chitosan, caused by the rupture of the glycosidic linkages between the glucosamine and N-acetylglucosamine rings [49, 50]. The comparison of data presented in Table 2 clearly shows that addition of filler to chitosan matrix caused lowering of thermal stability of samples. For CHT/5 C II, the 10% mass loss took place at temperature 18 °C lower than for CHT and the other two samples containing CNC I and C I behaved similarly. However, at 10% mass loss, there was almost no difference between chitosan sample (237 °C) and CHT/5 CNC II (236 °C). The tendency that composites filled with CNC II were characterized with lowest impairment of thermal stability was maintained throughout further measurement range. At 50% mass loss, it is obvious that discrepancy between pristine chitosan and its composites is definitely vaster but CHT/5 CNC II film stands out again. Temperature at which 50% mass loss was reached for composite with CNC II was 554 °C (672 °C for unmodified chitosan), while an average temperature for other films was in range of 424–450 °C.
More detailed look at TG results leaves no doubt that the thermal properties of composite films are highly dependent on the characteristics of (nano)metric fillers. Thus, below we will try and correlate them.
The TG results clearly show that sample containing CNC II was characterized with better thermal stability than film with CNC I. But then, composites with CNC II turned out to be more thermally stable than those with C II. Similar tendency was noted for composites based on (nano)cellulose I, but only if high mass loss is considered. It seems that not only polymorphic variety but also size of filler is important in terms of thermal stability. Naturally, a question for the reason behind this observation arises. It is known that during mercerization parallel chain arrangement of the cellulose I changes into more stable, antiparallel arrangement of cellulose II and more interplane hydrogen bonds are being formed. As said before, these bonds are important in terms of successful ionic liquid treatment, but additionally, they play an important role during the thermal degradation of the sample. In comparison with interchain bonds in C I, more energy is required to disrupt inter- and intrachain bonds in C II and therefore start the thermal decomposition process [27]. That is concluded to be the cause of better thermal stability of composites based on mercerized (nano)cellulose. The answer to why CNC II underwent degradation at higher temperatures than C II is not straightforward. It is widely accepted that small particles with large number of free-end chains present in the sample induce decomposition at lower temperature [51, 52]. According to that theory, TG results obtained for CHT/5 C II sample should be better than for CHT/5 CNC II. On the contrary, thermal stability of film with CNC II was the least impaired, and there was a strong resemblance between TG curve of CHT/5 CNC II and CHT sample. Probably, nanometric particles of CNC II were better incorporated in chitosan matrix and surrounded by it more tightly. The CNC I filler consisted of larger amount of micrometric particles (4.4% of the CNC II and 36.8% of CNC I with diameters ≥3 µm) and therefore could not have been so good wetted by chitosan. Consequently, they were less susceptible to thermal decomposition than particles of CNC I. Even if nanofiller had more free-end chains, at this level of loading it was not a leading issue. This idea is supported by work of Crews et al. [53] who related poor dispersion of filler in matrix with the lack of the hydrolysis being performed. The ionic liquid treatment of cellulose causes the disruption of some hydrogen bonds that are partially responsible for nonhomogenous structure of composites. Although C II particles had bigger diameters, presumably they were poorer bonded with chitosan matrix than CNC II. It is likely that improvement of thermal stability could be noticed if the addition of CNC to matrix would be higher. That was reported in paper [40] where the thermal stability of the chitosan-based films increased substantially with the incorporation of the nanometric cellulose.
Some published works support thesis that nanometric cellulose can effectively improve thermal stability of polymers [54, 55], but there are also studies that report no impact or even reduction in thermal stability [56]. In this paper, we did not succeeded to produce CNC-based composites with enhanced thermal stability, but we were able to correlate the type of cellulosic filler with thermal stability of the composites.
Mechanical and morphological properties of composites
Majority of scientific publications regarding nanometric cellulose points out its unique mechanical properties. The influence of nanometric cellulose on tensile properties of composites can be obtained even at its low concentrations. Our findings strongly support this hypothesis.
Table 3 presents tensile strength (TS), elongation at break (EB), and Young’s modulus (YM) parameters obtained for tested films.
Nanometric cellulose is well known for enhancement of mechanical properties of composites, even at very low loadings. Its unique mechanical properties were observed also in our composites. Addition of 1 and 5% of CNC I caused the value of YM to increase almost four times in relation to chitosan. For chitosan loaded with 3% of CNC I, this change was even more considerable—YM increased almost eight times, reaching the value of 1253 MPa. It is known that as the amount of the filler in polymer matrix increases, the gradual increase in Young’s modulus is observed. However, after specific optimal concentration was reached, YM decreased. It is related to the agglomeration of small particles and formation of bigger agglomerates that are no longer effective in terms of stress transfer. Therefore, for composites containing 5% CNC I, decrease in the YM value was observed. Similar relationship was reported for chitosan filled with unzipped multiwalled carbon nanotube oxides and agar/nanocrystalline cellulose composites [57, 58]. Accordingly, it seems that in case of CNC I the optimal amount of filler was not 1, but 3%. Though, when compared to composites filled with CNC II, these values become less impressive. It is so because addition of only 1% of CNC I was responsible for the increase in YM up to 3316 MPa. When the amount of CNC II in chitosan matrix increased, the value of YM decreased, but still were higher than for neat chitosan and CHT/CNC I composites.
In terms of TS enhancement 1% filler loading turned out to be the most effective. When compared to chitosan (25 MPa), the biggest increase in this parameter was measured for CHT/1 CNC II (80 MPa) and then for CHT/1 CNC I (47 MPa). As the amount of filler reached 3%, the TS for CHT/CNC I (27 MPa) was comparable to neat chitosan, whereas for CHT/CNC II it was still definitely higher (39 MPa). The lowest values of TS were noted for the highest, 5% addition of filler, and, taking into consideration standard deviation, these numbers were similar to the TS of chitosan.
The differences in the values of the EB parameter of the evaluated samples were quite vast. As expected, film of pristine chitosan was found to be the most elastic. In general, addition of filler causes the decrease in this parameter, and this thesis was also compliant with our results. Surprisingly, while all the other composites were characterized with reduced elongation, value obtained for CHT/1 CNC II was analogous to unmodified chitosan (36 and 42%, respectively).
SEM microphotographs of composites films presented in Fig. 6 show that even at the highest loading of filler (Fig. 6b, d) the homogeneity of samples was quite good. Considering samples with the lowest amount of filler (Fig. 6a, c), it is obvious that in CHT/1 CNC II film more particles of filler were observed. It can be also noticed that apart from some agglomeration of filler taking place, nanometric particles were still present in the analyzed samples.

SEM microphotographs for chitosan films with: a 1% CNC I, b 5% CNC I, c 1% CNC II, and d 5% CNC II
Atef et al. [58] prepared agar films with addition of 2.5, 5, and 10% of CNC. Composites with 2.5% of CNC were characterized with improved values of YM and TS, but at higher concentration of filler, an impairment was observed. At 2.5% CNC loading, EB parameter was lower than for unmodified agar. The increase in these tensile properties (YM, TS) was ascribed to the similar structure of CNC and chitosan which both have polysaccharide structure. Alternatively, lowering the mechanical properties with increasing loading of filler was a consequence of agglomeration of CNC and heterogeneous size distribution. Noted decrease in EB is characteristic for nanocomposites and is a result of rigid nature of the filler and strong interactions between components of composite, responsible for restriction of the matrix motion [59, 60]. Not only Atef et al. but other research groups [61, 62] too showed that due to good dispersion, low content of nanofiller (CNC) in polymer matrix (polylactide) is the best in terms of enhancement of mechanical properties. This hypothesis is also supported by this and our previous work [28, 40], but stays in opposite to Khan et al. [63] who claimed that 5% of CNC is optimal for chitosan. It comes as no surprise that aspect ratio of a filler, its interaction with polymer matrix, and uniform dispersion of filler are the main factors limiting mechanical properties of composites [63]. However, in this research, a polymorphic form of nanofiller seems to be the key variable.
As previously stated, the greatest enhancement of tensile properties was obtained for composite with 1% of CNC II as a filler. Decrease in EB parameter was also definitely smaller for this sample than for any other. The reason for such differences between CNC I and CNC II is explained below. During the mercerization of C I, formation of more uniform and also smoother particles of C II takes place. In terms of cellulosic fibers, mercerization process helps to relieve some residual stresses, what makes the fibers stronger [64]. The positive role of NaOH treatment of cellulosic materials used in composites was confirmed by Ichazo et al. [65] and Borysiak [25]. Similarly, composites of poly(ethylene oxide) with two polymorphic forms of cellulose nanocrystals (CNC) showed that the enhancement of tensile properties for composites with CNC II was greater than for CNC I [27]. It was caused by good interaction between polymer matrix and cellulose crystals which was determined by the hydrogen bonding. The same mechanism can be found in our composites, where CNC II with larger amount of inter- and intraplane bonds was better bonded with chitosan matrix than CNC I. Apart from the optimal concentration of the filler, findings of this research stay in opposite to the mechanical results obtained for chitosan filled with acid-derived CNC. Tensile properties of chitosan/acid-derived CNC composites [28] indicate that addition of CNC I is more effective than CNC II. The reason for that is probably the fact that the acid-derived CNC I had rather fibrillar structure, while the sample of acid-derived CNC II consisted of spherical or irregularly shaped particles. In case of composites with fibrillar fillers, the stress transfer mechanism is different from this occurring in composites with particulate fillers [66], thus differences in mechanical properties of such materials are understandable. However, in this case it seems that the amount of micrometric particles present in filler was the factor influencing the mechanical properties of composites the most. Analysis of particle size distribution clearly shows that even the sample of CNC I consisted of larger number of smaller particles than CNC II, the difference in total volume of particles ≤220 nm is not so distinct (approximately 53% for CNC I and 50% for CNC II). Definitely bigger divergence can be observed when the micrometric size range is considered. In this range, there is more small particles of CNC II than those of CNC I. They are still in the size of microns, but in contrast to CNC I the overwhelming amount of particles had diameters below 3 µm (only 4.4% of the CNC II sample and 36.8% of CNC I had diameters over 3 µm). Authors of paper on reinforcing properties of silica in polypropylene composites [67] showed that the increase in particle size entail enhanced debonding of the filler from polymer matrix. In that research, filling polypropylene with 1 µm silica gave better results than in case of 3 µm particles, but it has to be mentioned that of course, the best results were obtained for the smallest particles. Additionally, it is due to the fact that for smaller particles with higher total surface area, the mechanism of stress transfer is more efficient. As a result, in samples containing small silica particles formation of holes and voids on the fracture surface of the composite did not appear.
Concluding, three main factors limiting the mechanical properties of our composites were: (1) polymorphism of cellulose that affects formation of hydrogen bonds between matrix and filler, (2) the content of the filler in composites, and (3) size of the filler. At high loading rates, a loss of mobility of the polymer chains occurs making it harder to adapt to deforming forces. In order to withstand them, particles of filler undergo debonding, leading to formation of holes, presence of which decreases the mechanical properties of the composite. This undesirable pulling out of filler escalates even more if particles of filler are large. Here, composites of CHT/1 CNC II containing the smallest amount of filler with the lowest micrometric fraction were found to be the most mechanically resistant.
Conclusions
In this research, cellulose I and cellulose II were subjected to ionic liquid treatment leading to production of nanometric celluloses. They were subsequently used for forming chitosan-based composites. Films of chitosan with nanocrystalline cellulose I and nanocrystalline cellulose II were characterized in terms of mechanical and thermal properties. This research shows that enhancement of properties of chitosan/CNC composites is a very complex issue and not only the amount of micro- and nanometric fraction of the filler, but also its polymorphic form has a great influence on final properties of such biocomposites. More detailed findings of this work can be shortly summarized as follows:
-
Nanometric celluloses were successfully obtained with use of a commercially available ionic liquid. However, mercerization preceding ionic liquid treatment was found to facilitate the intercalation of cation coming from ionic liquid. As a consequence, sample of CNC II consisted of smaller amount of micrometric particles than CNC I.
-
TG results show that incorporation of micro- and nanometric cellulose of each polymorphic form resulted in lowering the thermal stability of composites when compared with unmodified chitosan. Interestingly, thermal stability of composites containing CNC II was higher than film with CNC I. This was related to poorer dispersion of CNC I filler in the chitosan matrix. Film with CNC II was also more thermally stable than analogous composite with C II, which was probably caused by less durable bonding of big C II particles with the matrix.
-
In terms of enhancing mechanical properties, sample with 1% loading of CNC II was definitely the most effective. It was due to a low addition of the filler that did not restrain adaptation to deforming forces and small particles of CNC II which were well hydrogen bonded with the matrix and at the same time were not prone to debonding from the chitosan matrix.
References
Wertz JL, Bédué O, Mercier JP. Cellulose science and technology. Lausanne: EPFL Press; 2010.
Kümmerer K, Menz J, Schubert T, Thielemans W. Biodegradability of organic nanoparticles in the aqueous environment. Chemosphere. 2011;82(10):1387–92. doi:10.1016/j.chemosphere.2010.11.069.
Cheng Q, Wang S, Rials TG, Lee SH. Physical and mechanical properties of polyvinyl alcohol and polypropylene composite materials reinforced with fibril aggregates isolated from regenerated cellulose fibers. Cellulose. 2007;14(6):593–602. doi:10.1007/s10570-007-9141-0.
Klemm D, Kramer F, Moritz S, Lindström T, Ankerfors M, Gray D, et al. Nanocelluloses: a new family of nature-based materials. Angew Chem Int Ed. 2011;50(24):5438–66. doi:10.1002/anie.201001273.
Brinchi L, Cotana F, Fortunati E, Kenny JM. Production of nanocrystalline cellulose from lignocellulosic biomass: technology and applications. Carbohyd Polym. 2013;94(1):154–69. doi:10.1016/j.carbpol.2013.01.033.
Van Den Berg O, Schroeter M, Capadona JR, Weder C. Nanocomposites based on cellulose whiskers and (semi)conducting conjugated polymers. J Mater Chem. 2007;17(26):2746–53. doi:10.1039/b700878c.
Samir MASA, Alloin F, Paillet M, Dufresne A. Tangling effect in fibrillated cellulose reinforced nanocomposites. Macromolecules. 2004;37(11):4313–6. doi:10.1021/ma035939u.
Beck-Candanedo S, Roman M, Gray DG. Effect of reaction conditions on the properties and behavior of wood cellulose nanocrystal suspensions. Biomacromolecules. 2005;6(2):1048–54. doi:10.1021/bm049300p.
Roman M, Winter WT. Effect of sulfate groups from sulfuric acid hydrolysis on the thermal degradation behavior of bacterial cellulose. Biomacromolecules. 2004;5(5):1671–7. doi:10.1021/bm034519+.
Li C, Wang Q, Zhao ZK. Acid in ionic liquid: an efficient system for hydrolysis of lignocellulose. Green Chem. 2008;10(2):177–82. doi:10.1039/B711512A.
Wasserscheid Peter WT. Ionic liquids in synthesis. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2003.
Mao J, Osorio-Madrazo A, Laborie M-P. Preparation of cellulose I nanowhiskers with a mildly acidic aqueous ionic liquid: reaction efficiency and whiskers attributes. Cellulose. 2013;20(4):1829–40. doi:10.1007/s10570-013-9942-2.
Xia S, Baker GA, Li H, Ravula S, Zhao H. Aqueous ionic liquids and deep eutectic solvents for cellulosic biomass pretreatment and saccharification. RSC Adv. 2014;4(21):10586–96. doi:10.1039/C3RA46149A.
Mao J, Heck B, Reiter G, Laborie MP. Cellulose nanocrystals’ production in near theoretical yields by 1-butyl-3-methylimidazolium hydrogen sulfate ([Bmim]HSO4)—mediated hydrolysis. Carbohyd Polym. 2015;117:443–51. doi:10.1016/j.carbpol.2014.10.001.
Man Z, Muhammad N, Sarwono A, Bustam M, Vignesh Kumar M, Rafiq S. Preparation of cellulose nanocrystals using an ionic liquid. J Polym Environ. 2011;19(3):726–31. doi:10.1007/s10924-011-0323-3.
Suzuki T, Kono K, Shimomura K, Minami H. Preparation of cellulose particles using an ionic liquid. J Colloid Interface Sci. 2014;418:126–31. doi:10.1016/j.jcis.2013.12.014.
Chowdhury S, Mohan RS, Scott JL. Reactivity of ionic liquids. Tetrahedron. 2007;63(11):2363–89. doi:10.1016/j.tet.2006.11.001.
Gräsvik J, Winestrand S, Normark M, Jönsson LJ, Mikkola JP. Evaluation of four ionic liquids for pretreatment of lignocellulosic biomass. BMC Biotechnology. 2014;14:34. doi:10.1186/1472-6750-14-34.
Lee SH, Doherty TV, Linhardt RJ, Dordick JS. Ionic liquid-mediated selective extraction of lignin from wood leading to enhanced enzymatic cellulose hydrolysis. Biotechnol Bioeng. 2009;102(5):1368–76. doi:10.1002/bit.22179.
Martins MAP, Frizzo CP, Moreira DN, Zanatta N, Bonacorso HG. Ionic liquids in heterocyclic synthesis. Chem Rev. 2008;108(6):2015–50. doi:10.1021/cr078399y.
Moniruzzaman M, Ono T. Separation and characterization of cellulose fibers from cypress wood treated with ionic liquid prior to laccase treatment. Biores Technol. 2013;127:132–7. doi:10.1016/j.biortech.2012.09.113.
Pernak J, Kordala R, Markiewicz B, Walkiewicz F, Popławski M, Fabiańska A, et al. Synthesis and properties of ammonium ionic liquids with cyclohexyl substituent and dissolution of cellulose. RSC Adv. 2012;2(22):8429–38. doi:10.1039/c2ra21502k.
Wang H, Gurau G, Rogers RD. Ionic liquid processing of cellulose. Chem Soc Rev. 2012;41(4):1519–37. doi:10.1039/c2cs15311d.
Trache D, Hussin MH, Haafiz MKM, Thakur VK. Recent progress in cellulose nanocrystals: sources and production. Nanoscale. 2017;9(5):1763–86. doi:10.1039/C6NR09494E.
Borysiak S. Influence of wood mercerization on the crystallization of polypropylene in wood/PP composites. J Therm Anal Calorim. 2012;109(2):595–603. doi:10.1007/s10973-012-2221-x.
Borysiak S. Fundamental studies on lignocellulose/polypropylene composites: effects of wood treatment on the transcrystalline morphology and mechanical properties. J Appl Polym Sci. 2013;127(2):1309–22. doi:10.1002/app.37651.
Yue Y, Zhou C, French AD, Xia G, Han G, Wang Q, et al. Comparative properties of cellulose nano-crystals from native and mercerized cotton fibers. Cellulose. 2012;19(4):1173–87. doi:10.1007/s10570-012-9714-4.
Borysiak S, Grząbka-Zasadzińska A. Influence of the polymorphism of cellulose on the formation of nanocrystals and their application in chitosan/nanocellulose composites. J Appl Polym Sci. 2016;133(3):42864. doi:10.1002/app.42864.
Kim Y, Jung R, Kim H-S, Jin H-J. Transparent nanocomposites prepared by incorporating microbial nanofibrils into poly(l-lactic acid). Curr Appl Phys. 2009;9(1):S69–71. doi:10.1016/j.cap.2008.08.010.
Nordqvist D, Idermark J, Hedenqvist MS, Gällstedt M, Ankerfors M, Lindström T. Enhancement of the wet properties of transparent chitosan—acetic-acid—salt films using microfibrillated cellulose. Biomacromolecules. 2007;8(8):2398–403. doi:10.1021/bm070246x.
Jaworska M, Kula K, Chassary P, Guibal E. Influence of chitosan characteristics on polymer properties: II. Platinum sorption properties. Polym Int. 2003;52(2):206–12. doi:10.1002/pi.1160.
Thakur VK, Thakur MK. Recent advances in graft copolymerization and applications of chitosan: a review. ACS Sustain Chem Eng. 2014;2(12):2637–52. doi:10.1021/sc500634p.
Li H, Du Y, Xu Y. Adsorption and complexation of chitosan wet-end additives in papermaking systems. J Appl Polym Sci. 2004;91(4):2642–8. doi:10.1002/app.13444.
Rad-Moghadam K, Dehghan N. Application of cellulose/chitosan grafted nano-magnetites as efficient and recyclable catalysts for selective synthesis of 3-indolylindolin-2-ones. J Mol Catal A Chem. 2014;392:97–104. doi:10.1016/j.molcata.2014.05.005.
Fernandes SCM, Freire CSR, Silvestre AJD, Pascoal Neto C, Gandini A. Novel materials based on chitosan and cellulose. Polym Int. 2011;60(6):875–82. doi:10.1002/pi.3024.
Dehnad D, Mirzaei H, Emam-Djomeh Z, Jafari S-M, Dadashi S. Thermal and antimicrobial properties of chitosan–nanocellulose films for extending shelf life of ground meat. Carbohyd Polym. 2014;109:148–54. doi:10.1016/j.carbpol.2014.03.063.
Diab MA, El-Sonbati AZ, Bader DMD. Thermal stability and degradation of chitosan modified by benzophenone. Spectrochim Acta Part A Mol Biomol Spectrosc. 2011;79(5):1057–62. doi:10.1016/j.saa.2011.04.019.
Puchalska A, Mucha M. Thermogravimetry of chitosan with nanofillers. Prog Chem Appl Chitin Deriv. 2011;XVI:31–42.
Swain SK, Dash S, Kisku SK, Singh RK. Thermal and oxygen barrier properties of chitosan bionanocomposites by reinforcement of calcium carbonate nanopowder. J Mater Sci Technol. 2014;30(8):791–5. doi:10.1016/j.jmst.2013.12.017.
Grząbka-Zasadzińska A, Smułek W, Kaczorek E, Borysiak S. Chitosan biocomposites based on enzymatically produced nanocrystalline cellulose. Eur Polym J (In review).
Dinand E, Vignon M, Chanzy H, Heux L. Mercerization of primary wall cellulose and its implication for the conversion of cellulose I → cellulose II. Cellulose. 2002;9(1):7–18. doi:10.1023/a:1015877021688.
Jiang G, Huang W, Wang B, Zhang Y, Wang H. The changes of crystalline structure of cellulose during dissolution in 1-butyl-3-methylimidazolium chloride. Cellulose. 2012;19(3):679–85. doi:10.1007/s10570-012-9689-1.
Remsing RC, Swatloski RP, Rogers RD, Moyna G. Mechanism of cellulose dissolution in the ionic liquid 1-n-butyl-3-methylimidazolium chloride: a 13C and 35/37Cl NMR relaxation study on model systems. Chem Commun. 2006;12:1271–3. doi:10.1039/B600586C.
Li Y, Liu X, Zhang S, Yao Y, Yao X, Xu J, et al. Dissolving process of a cellulose bunch in ionic liquids: a molecular dynamics study. Phys Chem Chem Phys. 2015;17(27):17894–905. doi:10.1039/c5cp02009c.
Mwaikambo LY, Ansell MP. Chemical modification of hemp, sisal, jute, and kapok fibers by alkalization. J Appl Polym Sci. 2002;84(12):2222–34. doi:10.1002/app.10460.
Gilbert RD, Kadla JF. Polysaccharides—cellulose. In: Kaplan DL, editor. Biopolymers from renewable resources. Berlin: Springer; 1998. p. 47–95.
Olsson A-M, Salmén L. The association of water to cellulose and hemicellulose in paper examined by FTIR spectroscopy. Carbohyd Res. 2004;339(4):813–8. doi:10.1016/j.carres.2004.01.005.
Kocherbitov V, Ulvenlund S, Kober M, Jarring K, Arnebran T. Hydration of microcrystalline cellulose and milled cellulose studied by sorption calorimetry. J Phys Chem B. 2008;112(12):3728–34. doi:10.1021/jp711554c.
Nam YS, Park WH, Ihm D, Hudson SM. Effect of the degree of deacetylation on the thermal decomposition of chitin and chitosan nanofibers. Carbohyd Polym. 2010;80(1):291–5. doi:10.1016/j.carbpol.2009.11.030.
Pereira FS, da Silva Agostini DL, Job AE, González ERP. Thermal studies of chitin–chitosan derivatives. J Therm Anal Calorim. 2013;114(1):321–7. doi:10.1007/s10973-012-2835-z.
Sofla MRK, Brown RJ, Tsuzuki T, Rainey TJ. A comparison of cellulose nanocrystals and cellulose nanofibres extracted from bagasse using acid and ball milling methods. Adv Nat Sci Nanosci Nanotechnol. 2016;7(3):035004.
Borsoi C, Zimmernnam MVG, Zattera AJ, Santana RMC, Ferreira CA. Thermal degradation behavior of cellulose nanofibers and nanowhiskers. J Therm Anal Calorim. 2016;126(3):1867–78. doi:10.1007/s10973-016-5653-x.
Crews K, Huntley C, Cooley D, Phillips B, Curry M. Influence of cellulose on the mechanical and thermal stability of ABS plastic composites. Int J Polym Sci. 2016;2016:10. doi:10.1155/2016/9043086.
Fernandes SCM, Freire CSR, Silvestre AJD, Pascoal Neto C, Gandini A, Berglund LA, et al. Transparent chitosan films reinforced with a high content of nanofibrillated cellulose. Carbohyd Polym. 2010;81(2):394–401. doi:10.1016/j.carbpol.2010.02.037.
Liu R, Tang C, Liu H. Cellulose nanofiber-protein composite. In: Pandey JK, Takagi H, Nakagaito AN, Kim H-J, editors. Handbook of polymer nanocomposites. Processing, performance and application: volume C: polymer nanocomposites of cellulose nanoparticles. Berlin: Springer; 2015. p. 449–64.
Silvério HA, Flauzino Neto WP, Pasquini D. Effect of incorporating cellulose nanocrystals from corncob on the tensile, thermal and barrier properties of poly(vinyl alcohol) nanocomposites. J Nanomater. 2013;2013:9. doi:10.1155/2013/289641.
Fan J, Shi Z, Ge Y, Wang Y, Wang J, Yin J. Mechanical reinforcement of chitosan using unzipped multiwalled carbon nanotube oxides. Polymer. 2012;53(2):657–64. doi:10.1016/j.polymer.2011.11.060.
Atef M, Rezaei M, Behrooz R. Preparation and characterization agar-based nanocomposite film reinforced by nanocrystalline cellulose. Int J Biol Macromol. 2014;70:537–44. doi:10.1016/j.ijbiomac.2014.07.013.
Azeredo HMC, Mattoso LHC, Avena-Bustillos RJ, Filho GC, Munford ML, Wood D, et al. Nanocellulose reinforced chitosan composite films as affected by nanofiller loading and plasticizer content. J Food Sci. 2010;75(1):N1–7. doi:10.1111/j.1750-3841.2009.01386.x.
Samir MASA, Alloin F, Paillet M, Dufresne A, Saïd Azizi MA. Tangling effect in fibrillated cellulose reinforced nanocomposites. Macromolecules. 2004;37(11):4313–6. doi:10.1021/ma035939u.
Ambrosio-Martín J, Lopez-Rubio A, Fabra MJ, Gorrasi G, Pantani R, Lagaron JM. Assessment of ball milling methodology to develop polylactide-bacterial cellulose nanocrystals nanocomposites. J Appl Polym Sci. 2015;132(10):41605. doi:10.1002/app.41605.
Haafiz MKM, Hassan A, Zakaria Z, Inuwa IM, Islam MS, Jawaid M. Properties of polylactic acid composites reinforced with oil palm biomass microcrystalline cellulose. Carbohyd Polym. 2013;98(1):139–45. doi:10.1016/j.carbpol.2013.05.069.
Khan A, Khan RA, Salmieri S, Le Tien C, Riedl B, Bouchard J, et al. Mechanical and barrier properties of nanocrystalline cellulose reinforced chitosan based nanocomposite films. Carbohyd Polym. 2012;90(4):1601–8. doi:10.1016/j.carbpol.2012.07.037.
Behery HM. Effect of mechanical and physical properties on fabric hand. 2005.
Ichazo MN, Albano C, González J, Perera R, Candal MV. Polypropylene/wood flour composites: treatments and properties. Compos Struct. 2001;54(2–3):207–14. doi:10.1016/S0263-8223(01)00089-7.
Gamstedt EK, Sandell R, Berthold F, Pettersson T, Nordgren N. Characterization of interfacial stress transfer ability of particulate cellulose composite materials. Mech Mater. 2011;43(11):693–704. doi:10.1016/j.mechmat.2011.06.015.
Omar MF, Akil HM, Ahmad ZA. Particle size—dependent on the static and dynamic compression properties of polypropylene/silica composites. Mater Des. 2013;45:539–47. doi:10.1016/j.matdes.2012.09.026.
Acknowledgements
This research was supported by the grant of Poznan University of Technology 03/32/DSPB/0703.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Grząbka-Zasadzińska, A., Amietszajew, T. & Borysiak, S. Thermal and mechanical properties of chitosan nanocomposites with cellulose modified in ionic liquids. J Therm Anal Calorim 130, 143–154 (2017). https://doi.org/10.1007/s10973-017-6295-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6295-3