
Abstract
Thermal decomposition of palladium acetylacetonate adsorbed on zinc oxide (ZnO), and the formation of palladium oxide (PdO) and palladium zinc alloy (PdZn) phases were studied. Two types of ZnO supports were prepared by the microemulsion method using different surfactants, i.e. hexadecyltrimethylammonium bromide (CTAB) and (Bis(2-ethylhexyl) sulfosuccinate sodium salt, AOT). The nanoparticles of ZnO synthesized in the presence of CTAB surfactant showed higher specific surface area, smaller crystallite size and more irregular shape. Palladium was loaded on the surface of obtained supports by the impregnation method from the acetone solution of palladium acetylacetonate. Thermogravimetric studies indicated that palladium precursor loaded on CTAB-modified ZnO support was less stable and simultaneously decomposed in broader range of temperatures. Slight differences between the forms of precursors adsorbed on the supports were demonstrated by the diffuse reflectance infrared Fourier transform spectroscopy studies. Thermal decomposition of palladium acetylacetonate precursors in the air led to the formation of PdO species. The influence of ZnO morphology on the metal–oxygen bonds strength in PdO and formation of active phases were observed. Strongly dispersed PdZn crystallites on ZnO supports were formed upon reduction at 350 °C. Smaller crystallites of the size equal to 6.5 nm were detected in the Pd/ZnO-AOT catalysts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Supported palladium catalysts have been widely used for oxidation and hydrogenation reactions of numerous chemical compounds [1, 2]. In recent years, a lot of attention has been paid to the development of supported palladium alloy catalysts, such as PdZn, PdIn or PdGa, showing their superior activity and selectivity in the hydrogenation of carbon oxides to methanol, water gas shift reaction, methanol steam reforming or hydrogenation of alkenes and alkadienes [3–7]. The impregnation method from aqueous solutions is one of simplest ways of the preparation of catalysts and has been often used for the synthesis of alumina-, silica- or carbon-supported palladium catalysts. It is well-accepted opinion that the support plays the crucial role in the formation of small crystallites and thus influences the activity, selectivity and durability of catalysts [8]. The formation of active sites in Pd/ZnO catalysts is a complex process and includes (1) adsorption of metal precursor on the support surface, (2) thermal decomposition of the metal precursor to oxide form (or directly to metallic form) and (3) reduction of oxide species to metallic palladium. The surface or bulk PdZn alloy phases are formed at elevated temperatures as a result of enhanced reduction of ZnO in the presence of metallic palladium. It has been often claimed that the size of PdO and Pd particles, PdO stability, interactions with the support and also catalytic performance of catalysts can be influenced by nature of palladium precursors [9–11]. Palladium nitrate Pd(NO3)2, tetraamminepalladium(II) nitrate Pd(NH3)4(NO3)2, tetraamminepalladium(II) chloride Pd(NH3)4Cl2 as well as palladium chloride PdCl2 belong to the most popular precursors used for preparation of catalysts, and their application in the impregnation method has been mostly determined by the isoelectric point (IEP) of the supports. Several papers demonstrated beneficial properties of palladium catalysts obtained by impregnation of the supports from nonaqueous solutions of organometallic complexes, such as palladium acetylacetonate (Pd(acac)2) [12, 13]. The use of aqueous solution may lead to the uncontrolled growth of palladium oxide species during drying and precursor decomposition. The way of adsorption of Pd(acac)2 on the oxide supports and transformation of the complex to palladium crystallites are still under debate. Palladium in the planar Pd(acac)2 complex is coordinated with two bidentate acetylacetonate ligands. β-diketonate ligands may interact with hydroxyl groups or defect sites on the supports, leading to the formation of hydrogen bonds or disruption of Pd-acetylacetonate ligand bonds with ligand exchange [14, 15]. There are a few works related to bulk and supported Pd(acac)2 decomposition [16–18]. Such phenomena have not been well documented for Pd/ZnO systems.
The properties of metal oxide supports can be modified by the changes of precipitation conditions, thermal treatment or introduction of secondary elements. The promising preparation method of ZnO with suitable structural and surface properties is precipitation in reverse microemulsion systems [19, 20]. The water-in-oil (w/o) microemulsion is composed of continuous phase—an organic solvent and dispersed phase—water solution containing metal salts or precipitants, which is stabilized by surfactants accumulated at the interphase region of micelles. The precipitation and growth of nanoparticles are controlled to some extend by the size or shape of micelles, related to the nature of surfactants, water to surfactant concentration and the presence of co-surfactants.
The general aim of our studies was to shed more light on the formulation of active Pd/ZnO catalysts for hydrogen production in the steam methanol reforming. In the present article, we have focused on the adsorption of Pd(acac)2 precursor, its decomposition and formation of PdZn alloys on two types of ZnO supports of different morphologies obtained by the application of microemulsion method.
Experimental
Synthesis of the supports and catalysts
Two types of zinc oxide supports were prepared in water-in-oil microemulsions, composed of cyclohexane (nonpolar phase), 0.5 M aqueous solutions of zinc nitrate or ammonium carbonate as precipitating agent (polar phase), n-butanol (co-surfactant) and cationic hexadecyltrimethylammonium bromide (CTAB) or anionic dioctyl sulfosuccinate sodium (Bis(2-ethylhexyl) sulfosuccinate sodium salt, AOT) surfactant, respectively. The water-to-surfactant molar ratio was 50, and the mass ratio of co-surfactant to surfactant was 1:1. The microemulsions containing zinc nitrate and ammonium carbonate were mixed and stabilized for 3 h at room temperature. Next, microemulsion was destabilized with tetrahydrofuran (THF) with volume three times greater than the volume of mixture. The obtained materials were centrifuged, transferred to filtration and washed with chloroform, water and ethanol. After 24 h, the samples were dried at 110 °C overnight and calcined at 400 °C for 6 h. The supports were denoted as ZnO-CTAB and ZnO-AOT, respectively. The obtained supports were impregnated with acetone solution of palladium acetylacetonate. After impregnation and mixing of the dispersed system for 2 h, the temperature was increased to 55 °C to complete acetone evaporation. The product was dried at 110 °C overnight and next calcined at 250 °C for 2 h.
Characterization
The nitrogen adsorption–desorption isotherms were obtained using ASAP 2405 (Micromeritics Instrument Corp.). The samples were outgassed at 200 °C. The BET method was used to calculate the specific surface area (S BET). The total pore volume (V p) was determined on the basis of nitrogen amount adsorbed at relative pressure of about p/p° = 0.98. Pore size distribution and mean pore diameter (D p) were estimated by the application of Barrett–Joyner–Halenda (BJH) method using the adsorption branch of isotherms.
Powder X-ray diffraction (XRD) studies were carried out in an Empyrean (PANalytical) diffractometer, using CuKα radiation (λ = 1.5418 Å). Diffraction patterns were used to phase identification and crystallite size calculation. The crystallite size was calculated from the broadening of the (101) XRD reflexion peak at 2θ = 36.2 using Warren–Scherrer equation:
where θ—diffraction angle, λ—X-ray wavelength (1.54179 Å for Cu lamp), B 1—full width at half maximum (FWHM) of the reflection, B 2—peak width of the standard.
The morphology of the samples was determined by the transmission electron microscopy. The samples were ground in an agate mortar to a fine powder. The resulting powder was poured with 99.8 % ethanol to form a slurry. The sample was inserted into the ultrasonic homogenizer for 20 s. After that, the slurry was pipetted and supported on a 200-mesh copper grid covered with lacey Formvar and stabilized with carbon (Ted Pella Company). The sample was then inserted to the holder and moved to the electron microscope. The electron microscope, Tecnai G2 20X-TWIN (FEI Company), equipped with LaB6 emitter was used. Microscopic studies of the samples were carried out at an accelerating voltage of the electron beam equal to 200 kV. The catalysts prior microscopic studies were reduced at 350 °C for 1 h in hydrogen.
Thermogravimetric studies were carried out by using the TG121 microbalance system (Cahn) under dynamic conditions. The dried sample (10 mg) was heated in airflow (70 cm3 min−1) with heating rate of 5 °C min−1.
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) studies were performed using Nicolet 6700 (Thermo Scientific) spectrometer equipped with LN2 cooled MCT/A detector. Samples were introduced into the Praying Mantis High Temperature Reaction Chamber (Harrick), which was covered by a dome with ZnSe windows, and were heated in the flow of air (30 cm3 min−1) with the heating rate of 5 °C min−1. DRIFTS spectra were collected at different temperatures with the resolution of 4 cm−1 and maximum source aperture. Interferograms of 256 scans were averaged for each spectrum. The spectra were corrected by applying the background signal recorded for KBr powder.
Temperature-programmed reduction (TPR) studies were carried out in the AutoChem II 2920 (Micromeritics Instrument Corp.). The sample (50 mg) was introduced into the quartz U-tube microreactor and was heated in the flow of H2 (5 vol %)/Ar mixture with the ramp rate of 10 °C min−1 from −30 to 600 °C.
Results and discussion
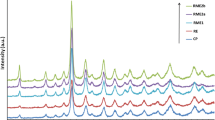
Microemulsion method was successfully used for preparation of zinc oxide supports of different structural and surface properties. The TEM images of the samples are presented in Fig. 1. The use of cationic surfactant (CTAB) led to the development of irregular, elongated ZnO nanoparticles (Fig. 1a). ZnO prepared in the presence of AOT (ZnO-AOT) contains more regular nanoparticles of flat walls and well-developed straight edges (Fig. 1b). The specific surface areas of obtained materials are equal to 22.3 and 12.8 m2 g−1, respectively (Table 1). Higher pore volume and mean pore diameter are observed for ZnO-CTAB support (Table 1). X-ray diffraction patterns of the samples are presented in Fig. 2. The XRD peaks well correspond to hexagonal ZnO phase (ICCD PDF4+ 04-009-7657). Mean crystallite size of ZnO-CTAB and ZnO-AOT is equal to 25.4 and 30.7 nm, respectively (Table 1). Small differences of the intensities of XRD peaks of nonpolar and polar planes could be observed. The ratio of intensities I (100)/(002) is equal to 1.25 for ZnO-CTAB and 1.35 for ZnO-AOT, respectively. The lower ratio I (100)/(002) means the higher proportion of surface sites located on nonpolar planes, which were formed along the c-axis [0001] direction during ZnO crystal growth [21].

Transmission electron microscopy images of the supports: a ZnO-CTAB, b ZnO-AOT

X-ray diffraction patterns of the supports
The supports were impregnated with acetone solution of palladium acetylacetonate. The results of thermogravimetric studies of decomposition of precursors in air after initial drying are presented in Fig. 3. The mass loss of Pd-loaded ZnO-CTAB sample becomes noticeable at about 130 °C. The maximum of DTG peak is located at the temperature of 180 °C. The observed changes are connected with decomposition of adsorbed Pd(acac)2 complexes and simultaneous oxidation of organic species. Bulk palladium acetylacetonate, according to the literature data, can be decomposed during heating in an inert gas or oxidizing atmosphere in the range 150–250 °C prior volatilization [16]. The course of thermal decomposition of supported Pd(acac)2 is sensitive to the gas phase composition. Studies presented in Ref. [22] indicated that decomposition of Pd(acac)2 adsorbed on SiO2 in oxidative atmosphere led to oxidation of organic ligand and formation of PdO particles. The decomposition of adsorbed precursor at 250 °C in inert atmosphere resulted in the formation of carbonaceous residues, which covered the surface of palladium crystallites. An increase in the thermal treatment of the samples to 500 °C facilitated reduction of palladium by carbon or hydrogen and transformation of carbonic material to the ‘carbide-like’ phases. Authors in Ref. [22] suggested that some residual could be very stable and hardly removed during reduction in hydrogen at 500 °C. Similar high stability of carbonaceous species remained on the surface of palladium nanoparticles was observed for organic palladium(II) complexes containing pyrazole ligands decomposed in inert gas [23, 24].

Thermogravimetric and derivative thermogravimetric curves recorded during thermal treatment of the supports loaded with palladium precursor in the flow of air
The irregular shape of DTG curve, presented in Fig. 3, reflects different thermal stability of precursors, resulted from the interactions of ligands with the support. The decomposition of precursor adsorbed on ZnO-AOT starts at slightly higher temperatures, at around 150 °C and proceeds initially almost in the same manner. The decomposition–oxidation peak in DTG curve for ZnO-AOT is much stronger. Its higher intensity is probably connected with palladium-catalysed oxidation of the traces of surface intermediates remained on the support after calcination. The maximum is located at 185 °C. Slight mass changes, observed in both samples at higher temperatures, in the range of 200–260 °C, can be ascribed to the decomposition–oxidation of surface zinc acetylacetonates.
Figure 4 shows DRIFTS spectra of bulk Pd(acac)2, ZnO supports before adsorption of the palladium salt, and the spectra of Pd-loaded ZnO supports recorded at different temperatures during thermal treatment in air. Low intensity of hydroxyl groups vibrations in high-wave number region (3800–3300 cm−1) and broad peaks in the range of 3500–2200 cm−1, which were observed for pure Pd(acac)2, indicate the presence of enolate form of acetylacetone ligands [25]. Chelate O-bonded form is also evidenced in the fingerprint region [26–27]. Strong IR vibrations observed in the range of 1600–1500 cm−1 can be attributed to the conjugated CO vibrations in a complex ring. The stretching vibrations of two interacting carbonyl groups in the keto form are usually situated above 1700 cm−1. Wide peak in the range of 1490–1325 cm−1 can be attributed to the asymmetrical and symmetrical deformation vibrations of CH3 groups [25]. The bands at 1358 and 1272 cm−1 are ascribed to the C–C stretching and C–C=C stretching in chelate ring vibrations. The in-plane C–H bending, CH3 rocking and C–CH3 stretching vibrations are located at 1199, 1023 and 937 cm−1, respectively.

Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectra of palladium acetylacetonate, the samples prior palladium precursor adsorption and recorded at different temperatures during thermal treatment in air: a Pd/ZnO-CTAB, b Pd/ZnO-AOT
Strong vibration bands of hydroxyl groups (in the range of 3800–3300 cm−1) and carbonate-like species (located in the range of 1690–1400 cm−1) are visible on the spectra of obtained ZnO supports. A slightly higher intensity of hydroxyl group vibrations in the range of 3800–3600 cm−1 can be found for Pd/ZnO-CTAB sample, indicating higher basicity of the solid surface. The decrease in the intensity of hydroxyl groups vibrations and shifts of the bands position towards lower wavenumbers is observed on the spectra of ZnO supports with Pd(acac)2 complex. This effect may indicate interaction of acetylacetonate ligands with hydroxyl groups. New bands are observed on the spectra of ZnO-CTAB sample after deposition of palladium acetylacetonate in the fingerprint region. The band at 1560 cm−1 and the shoulder at 1540 cm−1 are visible. Moreover, additional band appears at 1520 cm−1. C–H bending, CH3 rocking, C–C stretching and C–C=C stretching were also visible. Two distinct peaks within this region are observed for Pd/ZnO-AOT, almost at the same positions as for bulk Pd(acac)2. The disappearance of bands is observed with an increase in treatment temperature. Wide bands in the range of 1600–1400 cm−1 on the spectra of the samples heated at high temperatures in the flow of air suggest the presence of surface zinc carbonate phase. The negative charge in the enolate anions of β-diketones is delocalized over OCO atoms forming the chelate ring with palladium. Hence, the quasi-π-electron system of the acetylacetonates in planar complexes can directly interact with surface hydroxyl groups, leading to the formation of hydrogen bonds. Acetylacetonate complexes may also exhibit stronger specific interaction with the support through the coordinately unsaturated axial ligands of the metal ion [26]. These interactions may differ depending on the type of crystallographic planes of the supports and influence the thermal stability. We have observed that band positions of hydroxyl groups were located at higher wave numbers in ZnO-CTAB in comparison with ZnO-AOT. This can indicate different strengths of hydrogen bonds between acetylacetonate ligands and the support. Acetylacetonate ligands during adsorption and initial stages of decomposition may interact with the support forming zinc acetylacetonates; however, it is difficult to unambiguously state by DRIFTS the presence of such process.
The temperature-programmed reduction curves of the catalysts after calcination are presented in Fig. 5. The TPR studies proved that thermal treatment of Pd(acac)2 loaded on ZnO in the air led to the formation of palladium oxide. The samples show different reducibility. The reduction of PdO crystallites located on ZnO-CTAB support starts at very low temperature (−50 °C) and proceeds through mild maximum at 24 °C. The increase in signal intensity is connected with consumption of hydrogen in the reaction with PdO to Pd° and H2O [22]. Hydrogen at low temperatures is simultaneously dissolved in metallic palladium. The negative peak at 50 °C confirms β-PdH2 decomposition and desorption of H2 [22]. The reduction of PdO on ZnO-AOT begins at higher temperatures and goes through very rapid reduction maximum located at 42 °C. The negative peak was not observed for Pd/ZnO-AOT sample. Different routes of TPR curves point out on changes of palladium–oxygen interactions. These differences could be ascribed to the size or shape of crystallites, as well as to the interactions of PdO with the support.

Temperature-programmed reduction curves
The reduction curves show a broad peak at higher temperatures, attributed to the reduction of ZnO. This process is facilitated by metallic palladium, which simultaneously participates in the formation of PdZn alloys [3–5, 28–30]. It is interesting that reduction of ZnO and thus formation of PdZn start at lower temperature in Pd/ZnO-AOT sample. The maximum is observed at around 320 °C. The maximum of high-temperature reduction peak for PdO/ZnO-CTAB is located at 350 °C. The differences can be related to the size of palladium crystallites and thus phase boundary, as well as the nature of ZnO crystallographic planes [30].
TEM images of catalysts are presented in Fig. 6. One can find slight changes of the morphology of the supports. Palladium deposition on the surface of ZnO-CTAB and reduction of catalysts in hydrogen at 350 °C led to the decrease in the specific surface area of materials from 22.3 to 15.2 m2 g−1. The catalysts also show lower total pore volume and mean pore diameter (Table 1). Simultaneously, the mean crystallite size of ZnO is decreased from 25.4 to 24.4 nm. In the case of the ZnO-AOT support, the deposition of similar amounts of Pd and reduction in hydrogen resulted in the slight increase in S BET and increase in the total pore volume. ZnO nanoparticles, presented in Fig. 6b, seem to be less regular than in the ZnO-AOT support (Fig. 1b). The mean crystallite size of ZnO is decreased from 30.7 to 27.3 nm. X-ray diffraction studies of the catalysts after reduction are presented in Fig. 7. The ratio of I (100)/(002) crystallographic planes in ZnO of hexagonal structure, in spite of the surface and porosity changes, is nearly the same as for pure supports and equals 1.24 for Pd/ZnO-CTAB and 1.34 for Pd/ZnO-AOT, respectively. X-ray diffraction studies reveal that reduction of catalysts at 350 °C leads to the formation of the small PdZn alloy crystallites (PDF4+ 01-080-3086) (Fig. 7). Mean size of crystallites located on the ZnO-CTAB supports is slightly larger (7.3 nm) than in the Pd/ZnO-AOT catalyst (6.5 nm). TEM images evidenced very narrow crystallite size distribution. This is especially visible for Pd/ZnO-AOT catalyst. Nearly all species are located on the external surface of ZnO grains and could be easily accessible for substrates in the catalytic reactions.

Transmission electron microscopy images of the catalysts: a Pd/ZnO-CTAB, b Pd/ZnO-AOT

X-ray diffraction patterns of Pd/ZnO catalysts after reduction
Conclusions
Two types of zinc oxide supports were prepared by the microemulsion method using cationic and anionic surfactants: hexadecyltrimethylammonium bromide (CTAB) and (Bis(2-ethylhexyl) sulfosuccinate sodium salt, AOT), respectively. The obtained materials showed different structural and surface properties. The catalysts of 5 mass% of Pd were prepared by impregnation method using palladium acetylacetonate in acetone solution. The support influenced the way of adsorption of acetylacetonate precursor, thermal decomposition, reduction of PdO species and formation of alloy phases. Palladium precursor loaded on the CTAB-modified ZnO support was less stable and simultaneously decomposed in a more broad range of temperatures. Reduction of PdO phases in such catalysts started at the lower temperature, the maximum of reduction occurred at the lower temperature and intermediate β-PdH2 was observed. PdZn alloy was developed in the range of 250–400 °C. The alloy was formed more difficult in Pd/ZnO-CTAB catalysts, and this effect was mainly related to the larger size of palladium crystallites. Strongly dispersed PdZn crystallites on both types of supports were obtained. Smaller crystallites of the mean size equal to 6.5 nm were developed on the surface of ZnO nanoparticles prepared in the presence of AOT surfactant.
References
Centi G. Supported palladium catalysts in environmental catalytic technologies for gaseous emissions. J Mol Catal A Chem. 2001;173:287–312.
Teschner D, Révay Z, Borsodi J, Hävecker M, Knop-Gericke A, Schlögl R, Milroy D, Jackson SD, Torres D, Sautet P. Understanding Pd hydrogenation catalysts: when nature of the reactive molecule controls the nature of the catalyst active phase. Angew Chem. 2008;120:9414–8.
Tew MW, Emerich H, van Bokhoven JA. Formation and characterization of PdZn alloy: a very selective catalyst for alkyne semihydrogenation. J Phys Chem C. 2011;115:8457–65.
Iwasa N, Masuda S, Ogawa N, Takezawa N. Steam reforming of methanol over Pd/ZnO: effect of the formation of PdZn alloys upon the reaction. Appl Catal A Gen. 1995;125:145–57.
Chin YH, Dagle R, Hu J, Dohnalkova AC, Wang Y. Steam reforming of methanol over highly active Pd/ZnO catalyst. Catal Today. 2002;77:79–88.
Gao Z, Zhang Y, Li D, Werth CJ, Zhang Y, Zhou X. Highly active Pd–In/mesoporous alumina catalyst for nitrate reduction. J Hazard Mater. 2015;286:425–31.
Penner S, Lorenz H, Jochum W, Stöger-Pollach M, Wang D, Rameshan C, Klötzer B. Pd/Ga2O3 methanol steam reforming catalysts: part I. Morphology, composition and structural aspects. Appl Catal A Gen. 2009;358:193–202.
Albers P, Pietsch J, Parker SF. Poisoning and deactivation of palladium catalysts. J Mol Catal A Chem. 2001;173:275–86.
Polshettiwar V, Len C, Fihri A. Silica-supported palladium: sustainable catalysts for cross-coupling reactions. Coord Chem Rev. 2009;253:2599–626.
Machocki A, Rotko M, Gac W. Steady state isotopic transient kinetic analysis of flameless methane combustion over Pd/Al2O3 and Pt/Al2O3 catalysts. Top Catal. 2009;52:1085–97.
Wenge L, Deyong G, Xin X. Research progress of palladium catalysts for methane combustion. China Pet Process Pe. 2012;14:1–9.
Toebes ML, van Dillen JA, de Jong KP. Synthesis of supported palladium catalysts. J Mol Catal A Chem. 2001;173:75–98.
Lear T, Marshall R, Lopez-Sanchez JA, Jackson SD, Klapötke TM, Bäumer M, Rupprechter G, Freund HJ, Lennon D. The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J Chem Phys. 2005;123:174706.
Van Der Voort P, White MG, Vansant EF. Thermal decomposition of VO(acac)2 deposited on the surfaces of silica and alumina. Langmuir. 1998;14:106–12.
Van Veen JAR, Jonkers G, Hesselink WH. Interaction of transition-metal acetylacetonates with γ-Al2O3 surfaces. J Chem Soc Faraday Trans. 1989;85:389–413.
Janković B, Mentus S. Model-fitting and model-free analysis of decomposition of palladium acetylacetonate [Pd(acac)2]. J Therm Anal Calorim. 2008;94:395–403.
Daniell W, Landes H, Fouad NE, Knözinger H. Influence of pretreatment atmosphere on the nature of silica-supported Pd generated via decomposition of Pd(acac)2: an FTIR spectroscopic study of adsorbed CO. J Mol Catal A Chem. 2002;178:211–8.
Passonen VM, Semyannikov PP, Nazarov AS. Thermal decomposition of palladium acetylacetonate intercalated into dicarbon fluoride. Chem Sustain Dev. 2002;10:751–6.
Kołodziejczak-Radzimska A, Jesionowski T. Zinc oxide—from synthesis to application: a review. Materials. 2014;7:2833–81.
Sarkar D, Tikku S, Thapar V, Srinivasa RS, Khilar KC. Formation of zinc oxide nanoparticles of different shapes in water-in-oil microemulsion. Colloid Surf A. 2011;381:123–9.
Mclaren A, Valdes-Solis T, Li G, Tsang SC. Shape and size effects of ZnO nanocrystals on photocatalytic activity. J Am Chem Soc. 2009;131:12540–1.
Łomot D, Juszczyk W, Karpiński Z, Bozon-Verduraz F. Evolution of catalysts prepared from chlorine-free Pd/SiO2 precursors. J Chem Soc Faraday Trans. 1997;93:2015–21.
da Silva C, da Silva DAM, Rocha FV, Barra CV, Frem RCG, Netto AVG, Mauro AE, de Almeida ET. Synthesis, characterization, and thermal behavior of palladium(II) complexes containing 4-iodopyrazole. J Therm Anal Calorim. 2014;117:1327–34.
Díaz-Ayala R, Arroyo-Ramírez L, Raptis RG, Cabrera CR. Thermal and surface analysis of palladium pyrazolates molecular precursors. J Therm Anal Calorim. 2014;115:479–88.
Tayyari SF, Milani-nejad F. Vibrational assignment of acetylacetone. Spectrochim Acta A. 2000;56:2679–91.
Baltes M, Van Der Voort P, Weckhuysen BM, Rao RR, Catana G, Schoonheydt RA, Vansant EF. Synthesis and characterization of alumina-supported vanadium oxide catalysts prepared by the molecular designed dispersion of VO(acac)2 complexes. Phys Chem Chem Phys. 2000;2:2673–80.
Kenvint JC, White MG, Mitchell MB. Preparation and characterization of supported mononuclear metal complexes as model catalysts. Langmuir. 1991;7:1198–205.
Cubeiro ML, Fierro JLG. Partial oxidation of methanol over supported palladium catalysts. Appl Catal A Gen. 1998;168:307–22.
Wang Y, Zhang J, Xu H. Interaction between Pd and ZnO during reduction of Pd/ZnO catalyst for steam reforming of methanol to hydrogen. Chin J Catal. 2006;27:217–22.
Conant T, Karim AM, Lebarbier V, Wang Y, Girgsdies F, Schlögl R, Datye A. Stability of bimetallic Pd–Zn catalysts for the steam reforming of methanol. J Catal. 2008;257:64–70.
Acknowledgements
The research was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-06-024/09 Center of Functional Nanomaterials).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pawlonka, J., Gac, W., Greluk, M. et al. Application of microemulsion method for development of methanol steam reforming Pd/ZnO catalysts. J Therm Anal Calorim 125, 1265–1272 (2016). https://doi.org/10.1007/s10973-016-5525-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5525-4