
Abstract
The metal–organic frameworks of the formula Ln(bta)·nH2O where n = 3, 4, 5 or 6; bta = benzene-1,3,5-triacetate ion: [C9H9(COO)3]3−; Ln = La(III), Nd(III), Eu(III), Tb(III), Ho(III), Tm(III), Lu(III) were synthesized by the reaction of LnCl3·xH2O with ammonium salt of H3bta acid in water solution. Insoluble water compounds crystallize in the monoclinic or triclinic crystal system, and they are stable up to 30 °C. When heated, they decompose up to 160–200 °C in two stages to stable anhydrous compounds. The dehydrated compounds Ln(bta) are still crystalline and stable up to about 350 °C. When heated above 350 °C, they undergo decomposition where the intermediate products are Ln2O2CO3 or Tb4O6CO3 and the residues are: Ln2O3 or Tb4O7. The interpretation of IR spectra of title compounds was made taking into consideration the experimental and theoretical wavenumbers obtained for H3bta specie. The XPS spectra of H3bta and its complexes were recorded. The way of metal–carboxylate group of ligand coordination was discussed. This work presents the results of the XPS analyses, which allowed to determine the binding energies (E b) for the main lines of Ln3d, Ln4d, Ln5p, C1s, O1s spectra, spin–orbital splitting and full width at half maximum complexes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The field of inorganic open-framework materials is dominated by aminosilicates and phosphates, but metal coordination polymers based on organic ligands have emerged as an important family in the last two decades. These metal–organic frameworks (MOFs) are a class of crystalline hybrid materials in which metal ions in the solid structure are linked by various organic bridging ligands. As the bridging ligands in the MOF structures, there are used O- and N-donor ligands, but coordination centres are transition metal as well as lanthanide(III) ions [1, 2]. In that family of materials, the metal carboxylates are specially interesting since they contain both organic and inorganic components: they not only form open-framework structures resulting from the presence of the carboxylate function itself, but also where the carboxylate group acts as a linker between inorganic moieties. Lanthanide(III) carboxylates attract the attention because of their luminescence properties and their spectrally narrow emission [3–6]. The large magnetic moment of the most of the lanthanide(III) ions together with their anisotropy makes these ions very useful in preparation of magnetic materials [7, 8]. The compounds become interesting also in searching for promising application in drug delivery, gas absorption as well as separation and catalysis [9, 10].
Benzene multicarboxylate ligands are frequently applied in the construction of lanthanide(III) polymeric 1D, 2D and higher-dimensional frameworks. The exemplary compounds are lanthanide benzenedicarboxylates [11], benzene-1,3,5-tricarboxylates [12], benzene-1,2,4,5-tetracarboxylates [13] and benzene-1,2,3,4,5,6-hexacarboxylates [14]. In each of the series of lanthanide complexes, one can observe two or three isostructural series indicating that the decreasing lanthanide ion radius has influence on the complex structure.
Recently, some interest has arisen in the construction of lanthanide MOFs with flexible organic ligands for example: Tb(III) and Ho(III) phenylene-1,2-diacetates [15], Sm(III), Eu(III), Tb(III), Ho(III) phenylene-1,3-diacetates [16], lanthanide phenylene-1,4-diacetates [17] and benzene-1,3,5-triacetates (Scheme 1) [18–21] owing to their flexibility and conformational freedoms. The use of lanthanide ions in that construction is due to the high coordination number (8 or 9) as well as special properties of the lanthanide ions.

This paper describes the synthesis, structural and thermal characterization of selected lanthanide complexes with the flexible bta ligand. Taking into consideration the high but changing coordination number of Ln3+ ions, reducing of their radius, diversity of carboxylate group-Ln3+ binding mode [22] as well as the flexible character of bta ligand [23] which can adopt different conformations in the process of MOFs formation proceeded under the stable conditions of synthesis. The conditions of synthesis have great impact on the structure of MOFs [20]. The aim of this paper was also to determine the influence of temperature increase on the stability of crystalline networks of the complexes. Net water molecules in the complex structure should be easily removed from the outer- and inner- coordination spheres without the lanthanide–organic framework collapse. In this way, the coordinatively unsaturated lanthanide ions can cause catalytic or surface activity of the porous complex materials. This inspires the research groups to seek the porous lanthanide–organic frameworks (MOFs).
Experimental
Preparation of the lanthanide(III) complexes
The flexible bta ligand is versatile and can adopt different conformations in the formation of complexes so conditions of reaction have a great impact on the MOFs structure [20]. The whole series of lanthanide(III) complexes was prepared under exactly the same conditions: in the reaction of lanthanide(III) chlorides (pH 5.3–5.8) and the ammonium salt of H3bta (pH ≈ 5.5) in the 0.01 M solutions at 40 °C at the molar ratio 1:1. The LnCl3 solutions were obtained in the reactions of appropriate rare earth oxides with 2 M solution of HCl. Next, these LnCl3 solutions reacted with a soluble form of the ligand, which was ammonium salt bta(NH4)3. The precipitates were filtered, washed with hot water to remove NH4 + ions and dried at 30 °C to the stable mass. The complexes were identified by the method of elemental analysis, and the lanthanide contents were determined on the basis of the registered TG curves from the thermogravimetric analyses. The results of elemental analysis for the obtained complexes are gathered in Table 1.
Physical techniques
The complexes were identified by the method of elemental analysis with a CHN 2400 Perkin–Elmer analyser. The contents of metal ions were determined on the basis of the registered TG curves from the thermogravimetric analyses. The IR spectra of the compounds were recorded over the range 4000–400 cm−1 using a IR SPECORD M80 spectrophotometer. The thermal behaviour of the compounds was investigated by a Setsys 16/18 Setaram thermoanalyser, registering TG, DTG and DSC curves. The samples (7–8 mg) were heated in the ceramic crucible in the temperature range 30–1000 °C in the flowing air atmosphere (1 dm3 h−1) with the heating rate 10 °C min−1. Gaseous products of decomposition of Tb(III) complex were identified using a Netzsch TG 209 apparatus coupled to a Bruker FTIR 66 spectrophotometer. The samples were heated in the dynamic argon atmosphere with a rate of 20 °C min−1 up to 1000 °C using a ceramic crucible.
For studied complexes, there were conducted powder X-ray diffraction experiments (XRPD) at 30, 250 and 800 °C using a PANalytical Empyrean automated X-ray diffractometer with the XRK-900 RIGID high-temperature reaction chamber. Cu Kα radiation (λ = 1.54187 Å) was used for the diffraction experiments conducted in the non-continuous scanning mode. The first scan was at 30 °C; then, the sample was heated with the rate of 60 °C min−1 up to 250 °C and stabilized for 5 min and scanned using the goniometer in the theta–theta orientation, next heated up to 600 °C, stabilized again for 5 min and scanned and finally up to 800 °C in the same way. The samples were dried, fine-powdered and applied to a low-background sample holder. In a typical experiment, data were collected via continuous scan in the range of 2θ = 5–90° with the step size of 0.02626° and a scan time of 180 s per step.
The X-ray photoelectron spectra (XPS) were measured on a UHV Prevac spectrometer. Monochromatic Al Kα radiation (1486.6 eV) was used from an X-ray gun operated at 12 kV and 30 mA. The powder samples were pressed into pellets, and then, they were transferred via a sample probe in the electron energy analyser chamber which had a base pressure in the range of 10−11 mbar in the analyser. For all samples, the intense carbon 1s photopeak of aromatic hydrocarbon character in the compounds was used as a standard for the binding energy. The binding energy at the standard peak was determined to be 284.8 eV.
Computational details
The molecular structure optimization of the H3bta and corresponding vibrational harmonic frequencies were calculated using the DFT with B3LYP combined with 6-311++G(d,p) basis set using Gaussian 03 W program package [24]. The starting atomic coordinates were taken from the crystal structure of the H3bta (Scheme 2) determined from X-ray crystallography [23]. The absence of imaginary wavenumbers of the calculated vibrational spectrum confirms that the optimized structure of H3bta is stable. In order to get better correlation between the calculated and the experimental frequencies, the calculated vibrational frequencies were scaled by two different scaling factors, i.e. 0.983 up to 1700 cm−1 and 0.958 for greater than 1700 cm−1 [25]. The assignments of the calculated wavenumbers were examined by means of PEDs using VEDA 4 program [26].

Molecular structure of neutral H3bta [23]
Results and discussion
Thermal analysis
Thermogravimetric analyses (TG) were performed for dried, powdered, crystalline samples of the complexes in the range of 30–1000 °C under air as well as argon atmosphere. The TG data show that complexes of the general formula LnC9H9(COO)3·3–6H2O, where Ln = La(III), Nd(III), Eu(III), Tb(III), Ho(III), Tm(III), Lu(III) heated in air (Fig. 1) as well as argon atmosphere (Fig. 2), are stable up to 30 °C, and then, the dehydration process takes place.

TG, DTG and DSC curves of thermal decomposition of Tb(bta)·4H2O in air

TG and DTG curve of Tb(III) complex in argon
The TG and DSC curves show two steps of dehydration which means that in the structure of complexes there are hydrogen- and coordination-bonded water molecules.
Determination of the number of water molecules in the outer- and inner- coordination spheres of compounds on the basis of TG and DSC analysis is sometimes possible, but in majority of cases removing hydrogen-bonded water causes total dehydration in one stage as it is in the case of La(III) complex. Neither on TG, DTG nor DSC curves, two peaks are observed although the complex contains two types of differently bonded water molecules. In the case of the other complexes, hydrogen-bonded water molecules are disconnected in the first step at lower temperatures: 110, 100, 90, 80, 100, 95 °C for Nd(III), Eu(III), Tb(III), Ho(III), Tm(III) and Lu(III) complexes, respectively, and then, the molecules of coordinated water are released at about 145, 130, 126, 120, 145 and 140 °C for Nd(III) and successive compounds. This two-step dehydration process is clearly visible for example on the TG, DTG and DSC curves for the Tb(III) complex in air (Fig. 1), in argon (Fig. 2) as well as on the IR spectra of gaseous products of decomposition at 90 and 126 °C (Fig. 3). The process of involving the water molecules observed on the TG curves is characterized by total mass loss in the range 10.63–21.82 % (Table 2). The values of dehydration energy calculated from the DSC curves are the sum of the enthalpy of disconnection of hydrogen- and coordination-bonded water molecules. These values increase with the increasing atomic number of Ln(III) ions, but generally the total energy of dehydration is in the range 28.55–35.78 kJ mol−1 H2O in the complexes.

Line plot of FT-IR spectra of the evolved gases for Tb(bta)·4H2O (a); three-dimensional FT-IR spectra of gaseous products of thermal decomposition of Tb(bta)·4H2O (b) in argon
After dehydration, the anhydrous compounds are stable up to about 300–350 °C. Further heating leads to the degradation of anhydrous compounds, which is accompanied by burning of organic ligand. The final products of decomposition are suitable lanthanide oxides.
The simultaneous FT-IR analysis of gaseous products of thermal decomposition was made for Tb(bta)·4H2O complex (Fig. 3). The results obtained from TG-FT-IR measurement confirm that the first loss of mass observed in the temperature range 30–98 °C is connected only with the removal of outer-sphere water molecules. Figure 3 shows the separated spectra of valence and deformation vibration of water molecules at 3750 and 1750 and 1900–1300 cm−1 [27], and the second part of water spectra appearing at the higher temperature, about 126 °C belonging to coordination water. Formation of the stable, anhydrous compound is confirmed by the lack of bands in the existing anhydrous, stable complex in the temperature range 130–300 °C. During further decomposition of the complex at higher temperatures CO2, CO and hydrocarbons are released. The carbon dioxide bands showed strong absorption peaks of stretching vibration at 2361 and 2330 cm−1 and the weak deformation peaks at 720 and 670 cm−1. The bands at 2180 and 2114 cm−1 belong to CO molecules. The stretching absorption bands at 2900 and 2800 cm−1, deformation mode at 1460 cm−1 and rocking vibration bands at 800 cm−1 appear due to the presence of hydrocarbons in the gaseous product of fragments of organic ligand decomposition [28].
The thermal stability of dehydrated complexes increases with the increasing atomic number of lanthanides, that is with the decreasing ionic radius of lanthanides. Ln2O3 and Tb4O7 are formed as the final products of decomposition.
XRD studies
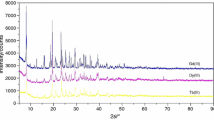
Bta is a flexible ligand on account of sp 3 carbon atoms (–CH2–) linking benzene as a central part of ligand with the carboxylic groups in the molecule [23]. Thus, this can cause that the series of complexes is entirely isostructural, in spite of significant differences in the size of the trivalent lanthanide ions, similar to phenylene-1,4-diacetates of lanthanides [17]. In the complexes, the lanthanide(III) ions are connected through oxygen atoms of carboxylate groups of organic ligand creating chains of metal ions. Part of water molecules completes the coordination number of lanthanide(III) ions to 9 for light and to 8 for heavy ones. Part of them is hydrogen bonded inside the complex structure. The analysed complexes crystallize in the triclinic or monoclinic crystal system (Table 3). As follows from the X-ray powder diffraction patterns (Fig. 4), the tetrahydrated ones are isomorphous. To examine stability of the crystalline frameworks of the complexes after removal of water molecules, the powder diffraction patterns of the compounds in situ at 30, 250 and 800 °C were recorded. As follows from the comparison of the diffractograms (Fig. 5), the same system of peaks is observed at 30 and 250 °C. This indicates that the framework and crystalline order are maintained despite removal of the water molecules [29]. The high stability of the anhydrous complexes frameworks is also reflected on the TG curves (Figs. 1, 2). The anhydrous compounds heated above 300–350 °C undergo decomposition where the intermediate products are Ln2O2CO3 and the residues are: Ln2O3 or Tb4O7 [30–33]. The above-mentioned final thermal decomposition products are confirmed by the data included in ICDD.

X-ray powder diffraction patterns of polycrystalline lanthanide(III) complexes

X-ray powder diffraction patterns of polycrystalline Ho(bta)·4H2O (30 °C), Ho(bta) (250 °C) and Ho2O3 (800 °C)
Infrared analysis
The infrared spectra of H3bta, its sodium salt and lanthanide(III) complexes were recorded, and assignments of bands occurring in the experimental as well as calculated spectra are gathered in Table 4. The exemplary experimental IR spectra of acid, its sodium salt and the Tb(III) and Ho(III) complexes are presented in Fig. 6. Comparing the IR spectra of acid and its metal compounds, one can observe that some bands characteristic of acid spectra disappeared in the spectra of sodium and lanthanide complexes. The stretching vibration bands of ν(OH) of –COOH groups exist as the broad band at 3500–2500 cm−1 in spite of hydrogen bonding in the acid structure. During complex formation, the strength of hydrogen bonding decreases and these bands are shifted to higher frequencies 3700–2700 cm−1. The ν(OH) band in the theoretical spectrum of acid is located at 3600 cm−1 (Table 4). The stretching asymmetric ν as(CH2) mode of methylene groups is observed in the experimental spectra at 2972, 2948 and 2970–2920 cm−1 at acid, sodium salt and Ln(III) complexes, but stretching symmetric ν s(CH2) vibrations are found at 2924, 2912 and 2920–2896 cm−1, respectively. The calculated values for these bonding vibrations are close to the experimental data, and they are within 2992–2905 cm−1. The deformation bands of scissoring vibrations δ(CH2) appear at 1385 cm−1 at acid (calcd. 1457, 1436 and 1435 cm−1), 1412 cm−1 at sodium salt and in the range of 1436–1416 cm−1 for the complexes; wagging mode ω(CH2) is located at 1266 cm−1 in the acid spectrum (calcd. 1287 cm−1), 1256 and 1272–1248 cm−1 for sodium and Ln(III) compounds. The twisting t(CH2) vibrations of methylene groups are seen at 1193 cm−1 for acid (calcd. 1194 and 1187 cm−1) at 1200, 1192 and 1192–1188 cm−1 for sodium and Ln(III) compounds, respectively. These bands do not shift due to complex formation because this part of ligand takes place neither in the metal coordination nor hydrogen bonding formation.

IR spectra of free H3bta acid (a), its sodium salt (b) and its Ho(III) (c) and Tb(III) (d) complexes
The carboxylic groups are the main groups in the structure of H3bta, which undergo transformation during deprotonation and metal complexes formation. The vibrational bands of carboxylic groups of acid contain the C=O, C–O and O–H vibration modes. The bands assigned to the stretching vibrations ν(C=O) of carboxylic group of acid molecule which usually appear about 1700 cm−1 in this acid appear as three strong peaks at 1723; 1705 and 1693 cm−1 (calcd. values 1743; 1741; 1733 cm−1) (Table 4; Fig. 6) due to small difference in the lengths of C=O bonds which depends on their environment in the acid structure. These peaks disappear in sodium salt and lanthanide complexes due to complete deprotonation of each carboxylic group and metal compound formation. So new, very strong bands assigned to the asymmetric ν as(COO−) and symmetric ν s(COO−) stretching vibrations of carboxylate anion appear. The asymmetric and symmetric bands of carboxylate group vibrations in sodium salt are located at 1580 and 1384 cm−1, whereas in the lanthanide complexes they are situated in the region of 1564–1536 and 1436–1396 cm−1 as presented in Fig. 6 and Tables 4 and 5. In all registered IR spectra of lanthanide(III) compounds, each absorption band of stretching symmetric carboxylate ν s(COO−) vibrations is split, which indicates the presence of more than one coordination mode of carboxylate groups of bta ligand. On the basis of the difference Δν between the wavenumbers ν as(COO−) and ν s(COO−) of carboxylate anion, the type of coordination of –COO− group by metal ions in the studied complexes can be analysed. According to the Nakamoto spectroscopic criterion [34], mode of carboxylate binding can be ionic, monodentate, bidentate chelating or bridging according to the series: Δν chelating < Δν bridging < Δν ionic < Δν monodentate. The Δν(COO−) for sodium salt is 196 cm−1, but is much lower in the series of lanthanide complexes (Table 5). Due to the above-mentioned rules, the obtained values of Δν(COO−) indicate the chelating, bridging and/or mixed chelating–bridging modes of the carboxylate groups in the investigated lanthanide(III) complexes (Scheme 1: mode b–h).
XPS spectroscopy
In order to determine electron redistribution between the acceptor and the donor related to complex formation, the XPS investigations were carried out. The indicator of these changes can be the difference in the binding energy (E b) of C1s and O1s electrons. All changes in binding energies of the electrons C1s and O1s in complexes spectra are the evidence of electron density redistribution in the O–C–O moiety caused by the complexation of the metal ions through the oxygen atoms of ligand. The difference between the E b C1s and O1s values of H3bta and its complexes can be the parameter that characterizes interactions of the carboxylate group with metal ions. This paper presents the results of the XPS analyses, which allowed to determine the binding energies (E b) for the main lines of C1s, O1s of H3bta as well as Ln3d, Ln4d, Ln5p, spin–orbital splitting and full width at half maximum (FWHM) [35] for lanthanide(III) complexes. The XPS data are listed in Tables 6–8. Figure 7 shows high-resolution XPS spectra of H3bta and lanthanide(III) complexes, and Fig. 8 shows deconvolution of only C1s and O1s spectra for H3bta and the exemplary Eu(bta)·4H2O complex. The deconvolution of the C1s spectrum of H3bta shows three peaks, each of them belongs to carbon atoms in different surrounding [36]. The most intense component with the lowest binding energy (284.7 eV) corresponds to aromatic carbon atoms of benzene ring, and its position is stable in the spectra of acid and all lanthanide(III) complexes. The second peak (286.1 eV) corresponds to carbons from the methylene groups being a link between the aromatic ring and the –COOH groups. The band with the highest energy of 289.2 eV corresponds to carbon atoms from the carboxylic groups of acid. There is no satellite peak associated with the shake-up effect.

High-resolution XPS spectra of H3bta and its complexes

C1s and O1s spectra of H3bta and Eu(bta)·4H2O
Table 6 and Fig. 8 demonstrate that position of E b (C–C) lines is stable in all spectra; the binding energies for electrons in the CH2 group in the complexes compared with that in H3bta are the same for the Nd(III), Tb(III), Ho(III) complexes and different for Eu(III) (ΔE 1 = 0.2 eV); and for La(III), Tm(III) and Lu(III) they cannot be extracted from C1s line. The main changes take place in the surrounding of carbon atoms which belong to the carboxylic groups. The binding energy of C1s decreases for the electrons in the O–C–O group in the complexes as compared to the O=C–O group in H3bta, in the range ΔE 1 = 0.3–0.5 eV. The change in the value of binding energy is an indicator of complex formation and an evidence of substantial variation of electron density in the carboxylate groups of different complexes. Decrease of E b means that the electron density on the atom increased upon complex formation. The deconvolution of the O1s spectrum of anhydrous H3bta shows two peaks. Both peaks O1s belong to the carboxylic group. First (532.3 eV) belongs to the carbonyl (C=O) oxygen atom and the second one (533.6 eV) to hydroxyl (OH) oxygen atom from carboxylic acid group. The formation of the complexes causes that energy binding of O1s electrons in the O–C–O groups of bta ligand becomes equal [36]. In the case of analysed complexes, one intensive line about 532 eV and the second one above 533 eV of low intensity can be observed instead of two intensive bands occurring in the spectrum of H3bta (Fig. 8). This single intensive line located around 532 eV corresponds to one type of oxygen atoms, namely oxygen atoms forming carboxylate groups. The single line O1s is shifted towards smaller E b values compared to acid spectrum, and the changes are in the range ΔE 2 = 0.2–0.5 eV (Table 6). The FWHM of the O1s line is indicative of either one state of the O atom or insignificant difference of energy between two states, which makes them almost identical. The second peak of E b equal 533.5 eV corresponds to the oxygen atom derived from water molecules [37]. All changes in binding energy C1s and O1s between H3bta and complexes are the evidence of the redistribution of electron density in the O–C–O moiety caused by the complexation of the metal through the oxygen atoms. Figure 9 shows the deconvolution of La3d spectrum with four lines corresponding to spin–orbit splitting (La3d3/2 and La3d5/2) and their shake-up satellites at higher binding energy side. The energy separation between main lines ΔE3 (3d3/2–3d5/2) and the satellite lines ΔE 4 (3dsat.–3d3/2 and 3dsat.–3d5/2) is 16.7, 3.3 and 3.2 eV, respectively. Satellites are assigned to charge-transfer shake-up from ligand to empty 4f orbital [38–41]. One peak corresponds to the final state without the charge transfer 4f0, and the satellite peak corresponds to the final state after transfer of electron (4f1) from ligand to 4f orbital of Ln(III). In Tb(III) complex, spin–orbit splitting (3d3/2 and 3d5/2) and satellites are also observed. Energy separation between the main lines ΔE3 and the satellite lines ΔE4 is 34.7, 6.7 and 9.2 eV, respectively. The values ΔE4 are different; in the case of La(III) compound, there is a small difference, but for Tb(III) complex it is larger. In the case of Nd(III) and Eu(III) complexes, only spin–orbit splitting lines are observed. An increase in the atomic number is accompanied by an increase in the binding energy 3d, 4d, 5p, and separation energy is shown in Fig. 10. All data are summarized in Table 8.

3d XPS spectrum of La(bta)·6H2O

Energy binding of 3d, 4p, 4d, 5p XPS lines and energy separation of 3d lines as a function of atomic number Z
Conclusions
The obtained lanthanide(III) complexes of the formula LnC9H9(COO)3·3–6H2O, where Ln = La(III), Nd(III), Eu(III), Tb(III), Ho(III), Tm(III) and Lu(III), are crystalline powders crystallizing in the monoclinic or triclinic crystal system. The complexes are stable under normal conditions, and removal of all water molecules does not lead to framework collapse. When heated, they decompose up to about 160–200 °C in two stages to stable anhydrous complexes. This indicates that in the structure of complexes there are hydrogen- and coordination-bonded water molecules. The values of dehydration energy increase with the increasing of atomic number of Ln(III) from 28 to 35 kJ mol−1 H2O. The XRPD investigations of the compounds indicate that the framework and crystalline order are maintained despite removal of water molecules. The framework of anhydrous Ln(bta) is highly stable, but heated above 300–350 °C undergoes decomposition to Ln2O3 or Tb4O7. On the basis of IR spectra, the different ways of metal ions coordination through carboxylate oxygen atoms are possible: chelating, bridging and/or bridging–chelating. The XPS electronic spectra confirmed the deprotonation of carboxylic groups of H3bta and their full transformation into carboxylate systems during complexes formation. The XPS study also revealed that in the structure of analysed complexes water molecules are present.
References
Rao CNR, Natarajan S, Vaidhyanathan R. Metal carboxylates with open architectures. Angew Chem Int Ed. 2004;43:1466–96.
Rao CNR. Chemistry of materials. A letter to a young friend. Resonance. 2009;14:508–17.
Allendorf MD, Bauer CA, Bhakta RK, Houk RJT. Luminescent metal–organic frameworks. Chem Soc Rev. 2009;38:1330–52.
Parra DF, Forster PL, Łyszczek R, Ostasz A, Lugao AB, Rzączyńska Z. Thermal behavior of the highly luminescent poly(3-hydroxybutyrate): Eu(tta)3(H2O)2 red-emissive complex. J Therm Anal Calorim. 2013;114:1049–56.
Łyszczek R. Hydrothermal synthesis, thermal and luminescent investigations of lanthanide(III) coordination polymers based on the 4,4′-oxybis(benzoate) ligand. J Therm Anal Calorim. 2012;108:1101–10.
Łyszczek R, Ostasz A, Bartyzel A, Lipke A. Thermal, spectroscopic and luminescence investigations of lanthanide(III) coordination polymers based on V-shaped 4,4′-sulfonyldibenzoic acid. J Anal Appl Pyrolysis. 2015;115:370–8.
Kurmoo M. Magnetic metal–organic frameworks. Chem Soc Rev. 2009;38:1353–79.
Li M, Liu B, Wang B, Wang Z, Gao S, Kurmoo M. Erbium-formate frameworks templated by diammonium cations: syntheses, structures, structural transition and magnetic properties. Dalton Trans. 2011;40:6038–46.
Hawes CS, Chilton NF, Moubaraki B, Knowles GP, Chaffee AL, Murray KS, Batten SR, Turner DN. Coordination polymers from a highly flexible alkyldiamine-derived ligand: structure, magnetism and gas adsorption studies. Dalton Trans. 2015;44:17494–507.
Wang LL, Luo F, Dang LL, Li JQ, Wu XL, Liu SJ, Luo MB. Ultrafast high-performance extraction of uranium from seawater without pretreatment using an acylamide- and carboxyl-functionalized metal–organic framework. J Mater Chem A. 2015;3:13724–30.
Jin HG, Yan YZ, Li J, Gu ZG, Chen JH, Liu YT, Zheng ZP, Zhan QG, Cai YP. 1-D to 3-D lanthanide coordination polymers constructed from 5-aminoisophthalic acid and oxalic acid. Inorg Chem Commun. 2012;23:25–30.
Liu K, You H, Jia G, Zheng Y, Song Y, Yang M, Huang Y, Zhang H. Coordination-induced formation of one-dimensional nanostructures of europium benzene-1,3,5-tricarboxylate and its solid-state thermal transformation. Cryst Growth Des. 2009;9:3519–24.
Rzączyńska Z, Ostasz A, Sikorska-Iwan M, Głuchowska H, Olszewska E, Pikus S. Synthesis and characterization of metal polycarboxylates constructed from lanthanides(III) and 1,2,4,5-benzenetetracarboxylic acid. J Therm Anal Calorim. 2006;84:575–9.
Ostasz A, Łyszczek R, Mazur L, Sienkiewicz-Gromiuk J, Rusinek I, Rzączyńska Z. Some properties of Nd and Er complexes with 1,2,3,4,5,6-benzeneheksacarboxylic (mellitic) acid. J Anal Appl Pyrolysis. 2013;99:203–10.
Li X, Wang C, Zheng X, Zou Y. Hydrothermal synthesis and crystal structure of two new lanthanide coordination polymers with 1,2-phenylenediacetate. J Coord Chem. 2008;61:1127–36.
Ren YX, Zhang ML, Li DS, Fu F, Wang JJ, Du M, Hou XY, Wu YP. Unusual (4,6)-connected lanthanide 1,3-phenylenediacetate coordination frameworks displaying lanthanide contraction effect and 1D and 2D mixed self-weaving architecture. Inorg Chem Commun. 2011;14:231–4.
Rusinek I, Sienkiewicz-Gromiuk J, Mazur L, Rzączyńska Z. Lanthanide(III) 1,4-phenylenediacetate complexes: the relation between the structure and thermal properties. J Inorg Organomet Polym. 2013;23:1068–77.
Zhang ZH, Shen ZL, Okamura T, Zhu HF, Sun WY, Ueyama N. Synthesis and structures of two series of coordination frameworks based on the assembly of 1,3,5-benzenetriacetic acid with lanthanide metal salts. Cryst Growth Des. 2005;5:1191–7.
Zhang ZH, Liu GX, Okamura T, Sun WY, Ueyama N. Synthesis, structures, and magnetic properties of new rare earth coordination polymers constructed by 1,3,5-benzenetriacetic acid. Struct Chem. 2006;17:3–11.
Zhang ZH, Meng WL, Okamura TA, Kong LY, Sun WY, Ueyama N. Lanthanide-organic framework with flexible triacid ligand: structural variation under different reaction conditions. Supramol Chem. 2006;18:317–25.
Zhang ZH, Okamura T, Hasegawa Y, Kawaguchi H, Kong LY, Sun WY, Ueyama N. Syntheses, structures, and luminescent and magnetic properties of novel three-dimensional lanthanide complexes with 1,3,5-benzenetriacetate. Inorg Chem. 2005;44:6219–27.
Huang YG, Jiang FL, Yuan DQ, Wu MY, Gao Q, Wei W, Hong MC. Intricate 3D lanthanide-organic frameworks with mixed nodes nets. J Solid State Chem. 2009;182:215–22.
Fitzgerald LJ, Gerkin RE. Interwoven hydrogen-bonding networks in benzene-1,3,5-triacetic acid. Acta Crystallogr C. 1997;53:967–9.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Ciosłowski J, Fox DJ. Gaussian 03 W: Revision D.1. Gaussian Inc. Wallingford; 2005.
Cinar M, Karabacak M, Asiri AM. An experimental and density functional study on conformational and spectroscopic analysis of 5-methoxyindole-2-carboxylic acid. Spectrochim Acta A. 2015;137:670–6.
Jamróz MH. Vibrational energy distribution analysis: VEDA 4.0 program. Warsaw; 2004.
Silverstein R, Webster FX. Spectrometric identification of organic compounds. New York: Wiley; 1998.
Łyszczek R, Mazur L. Synthesis, crystal structure, spectroscopic and thermal investigations of neodymium(III) biphenyl-4,4′-dicarboxylate framework. Cent Eur J Chem. 2012;10:1165–74.
Kustaryono D, Kerbellec N, Calvez G, Freslon S, Daiguebonne C, Guillou O. New family of porous lanthanide-containing coordination polymers: [Ln2(C2O4)3(H2O)6, 12H2O]∞ with Ln = La–Yb or Y. Cryst Growth Des. 2010;10:775–81.
Łyszczek R. Synthesis, structure, thermal and luminescent behaviors of lanthanide—pyridine-3,5-dicarboxylate frameworks series. Termochim Acta. 2010;509:120–7.
Rzączyńska Z, Kula A, Sienkiewicz-Gromiuk J, Szybiak A. Synthesis, spectroscopic and thermal studies of 2,3-napthalenedicarboxylates of rare earth elements. J Therm Anal Calorim. 2011;103:275–81.
Sienkiewicz-Gromiuk J, Rzączyńska Z. Structural, thermal, and spectra investigations of the lanthanide(III) biphenyl-4,4′-dicarboxylates. J Therm Anal Calorim. 2013;112:877–84.
Rzączyńska Z, Sienkiewicz-Gromiuk J, Głuchowska H. Structural and thermal properties of rare earth complexes with 2,2′-biphenyldicarboxylic acid. J Therm Anal Calorim. 2010;101:213–9.
Nakamoto K. Infrared and Raman spectra of inorganic coordination compounds. New York: Wiley; 1968.
Akitsu T, Einaga Y. Structures, magnetic properties and XPS of cyanide-bridged NdIII/SmIII/GdIII–CrIII complexes. Inorg Chim Acta. 2006;359:1421–6.
Stevens JS, de Luca AC, Pelendritis M, Terenghi G, Downes S, Schroeder SLM. Quantitative analysis of complex amino acids and RGD peptides by X-ray photoelectron spectroscopy (XPS). Surf Interface Anal. 2013;45:1238–46.
Ivanova TM, Maslakov KI, Linko RV, Evstifeev IS, Kiskin MA, Shcherbakov IN, Savilov SV, Lunin VV, Novotortsev VM, Eremenko IL. XPS study of the electronic structure of heterometallic complexes Fe2MO(O2CCH3)6(H2O)3]·3H2O (M = Co, Ni). Russ J Inorg Chem. 2014;59:670–7.
Ivanova TM, Kochur AG, Maslakov KI, Kiskin MA, Savilov SV, Lunin VV, Novotortsev VM, Eremenko IL. XPS study of the electron structure of heterometallic trinuclear complexes Fe2M(µ3-O)(µ-Piv)6(HPiv)3 (M = Mn Co, Ni). J Electron Spectrosc Relat Phenom. 2015;205:1–5.
Lixin Z, Bo W, Shiyan Y, Daosen J. XPS study of metal complexes with an unsymmetrical tridentate Schiff base. Polyhedron. 1993;12:1607–11.
Mullica DF, Lok CKC, Perkins HO, Benesh GA, Young V. The X-ray photoemission spectra of Nd(OH)3, Sm(OH)3, Eu(OH)3 and Gd(OH)3. J Electron Spectrosc Relat Phenom. 1995;71:1–20.
Ivanova TM, Shcherbakov IN, Linko RV, Kiskin MA, Evstifeev IS, Sidorov AA, Novotortsev VM, Eremenko IL. XPS study of the electronic structure of heterometallic complexes Fe2MO(Piv)6(HPiv)3 (M = Ni, Co). Russ J Inorg Chem. 2013;58:945–50.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sienkiewicz-Gromiuk, J., Rusinek, I., Kurach, Ł. et al. Thermal and spectroscopic (IR, XPS) properties of lanthanide(III) benzene-1,3,5-triacetate complexes. J Therm Anal Calorim 126, 327–342 (2016). https://doi.org/10.1007/s10973-016-5521-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5521-8