
Abstract
New thermoplastic polyurethane elastomers (TPUs) were obtained by a one-step melt polyaddition from 30, 45 and 60 mol% aliphatic polycarbonate diol of M n = 2000 g mol−1 (Desmophen® C2200, Bayer), 1,1′-methanediylbis(4-isocyanatocyclohexane) (HMDI) or 1,6-diisocyanatohexane (HDI) and 2,2′-[sulfanediylbis(benzene-1,4-diyloxy)]diethanol (acting as a chain extender). The TPUs were examined by FTIR, UV–Vis, atomic force microscopy, X-ray diffraction analysis, differential scanning calorimetry, thermogravimetry (TG) and TG–FTIR. Moreover, their Shore A/D hardness, tensile, adhesive and optical properties were determined. The obtained TPUs were transparent or opaque high molar mass materials, showing amorphous or partially crystalline structures. The HDI-based TPUs exhibited lower glass-transition temperatures than those based on HMDI (from −35 to −31 °C vs. from −20 to 1 °C) as well as a higher degree of microphase separation. The TPUs were stable up to 262–272 °C, taking into account the temperature of 1 % mass loss, with somewhat higher values shown by those from HDI. They decomposed in two stages. The main volatile products of the hard-segment decomposition were carbon dioxide, water and carbonyl sulfide, while aliphatic ethers, aldehydes and unsaturated alcohols, as well as carbon dioxide, originated from the soft-segment decomposition. The synthesized TPUs, with hardness in the range of 80–90°Sh A, possessed a good tensile strength (33.0–38.7 MPa), similar to that of the commercial biodurable medical grade TPU ChronoFlex® AL 80A (37.9 MPa), prepared from aliphatic polycarbonate diol, HMDI and butane-1,4-diol.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Properties of polyurethanes can be widely adjusted by changing the composition of the raw materials and the processing conditions. Owing to that, polyurethanes constitute a group of polymers of most versatile properties and a very wide range of industrial application. They are used mainly as foams, elastomers, coatings, fibers and adhesives. Polyurethanes, especially thermoplastic polyurethane elastomers (TPUs), are now one of the fastest growing areas in polymer technology [1, 2].
The basic reagents used for the synthesis of TPUs, which are multiblock copolymers, are diisocyanates and short-chain diols that build hard segments and long-chain diols that constitute soft segments. Commercial TPUs are mainly synthesized from 1,1′-methanediylbis(4-isocyanatobenzene) (MDI), butane-1,4-diol (BD) as a chain extender and polyether, polyester or polycarbonate diols. In order to obtain TPUs resistant to UV radiation (non-yellowing), MDI is substituted by aliphatic diisocyanates (mostly 1,1′-methanediylbis(4-isocyanatocyclohexane) (HMDI), while in order to obtain polymers with higher modulus of elasticity and hardness as well as better thermal stability, both aliphatic–aromatic and aromatic “bulky” diols are used instead of BD. In general, polyester diols yield TPUs with higher tensile strength, harder and less extensible than polyether diols. Moreover, polyester TPUs show better oxidation stability but worse hydrolytic resistance as compared to polyether TPUs. TPUs based on polycarbonate diols (PCDs) combine excellent tensile strength with good oxidation and hydrolytic stability. Because of these advantageous properties, polycarbonate TPUs are widely used in many biomedical applications [2–5].
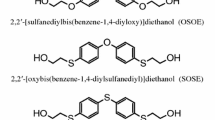
This study is a part of an ongoing investigation concerning new TPUs obtained from aliphatic–aromatic sulfur-containing chain extenders, derivatives of, among others, diphenylmethane, diphenylethane, benzophenone, diphenyl ether and diphenyl sulfide [6–13]. On the basis of the studies conducted for selected polymers, it was stated that the introduction of sulfur atoms to the polymer chain improved their adhesive strength [12, 14–16] and refractive index [15, 16]. Polymer antimicrobial activity against Gram-positive bacteria was also discovered [15]. The aim of the study was to synthesize and determine the structure and some properties of new TPUs based on a chain extender containing diphenyl sulfide units, i.e., 2,2′-[sulfanediylbis(benzene-1,4-diyloxy)]diethanol (diol OSOE), HMDI (Desmodur W®, Bayer) or 1,6-diisocyanatohexane (HDI) and 30, 45 and 60 mol% aliphatic PCD of M n = 2000 g mol−1 (Desmophen® C2200, Bayer). This relatively easy to obtain chain extender has already been applied by us for the synthesis of MDI-based TPUs with the polyether soft segment of M n = 1000 g mol−1. The polymers showed a relatively good microphase separation, thermal stability and tensile strength (up to ~44 MPa) [12]. Moreover, using this chain extender, a synthesis and characterization of HMDI- and MDI-based segmented polyurethanes from commercial poly(hexane-1,6-diyl carbonate) diol of M n = 860 g mol−1 were carried out, which exhibited high transparency (transmittance at 500 nm: 80–87 %, at 800 nm: 84–89 %), but unfortunately were not elastomers [unpublished data]. The main interest of this study was to obtain non-yellowing TPUs with good mechanical properties and relatively good thermal stability. Additionally, it was interesting to check the possibility of obtaining highly transparent polymers by using the above-mentioned commercial PCD of M n = 2000 g mol−1. Polymers showing a good transparency have already been synthesized by Da-Kong Lee et al. [17] from two PCDs (L6002 and L5652) of M n = 2000 g mol−1 supplied by the Asahi Kasei Corporation, MDI and BD. High transparency is advantageous and may enhance the product value of TPUs [17–19].
The present study determined the influence of the kind of diisocyanate used and the soft-segment content on the physicochemical, thermal and mechanical properties, as well as transparency of the resulting TPUs. Moreover, for selected TPUs, refractive index and adhesive properties were investigated. The work also gives a thermal characterization of the newly obtained regular polyurethanes (RPURs) based on diol OSOE and HMDI (HM) or HDI (H), building the hard segments in these TPUs.
For the TPUs, the system of designations was as follows (see Table 1): X–Y, where X is the abbreviations of the diisocyante and Y represents the soft-segment content. For example, H-30 is a polymer prepared from HDI and 30 mol% PCD.
Experimental
Materials
The diol OSOE (m.p. = 101–102 °C) after recrystallization first from methanol/water and next from 1,2-dichloroethane) was prepared from 4,4′-sulfanediyldiphenol and ethylene carbonate by a modified procedure described by Penczek and al. [20]. PCD (Desmophen® C2200) of \( \overline{M}_{\text{n}} \) = 2000 g mol−1 and HMDI (99.5 %, Desmodur W®) were kindly supplied from Bayer (Germany). Before being used, the PCD was heated at 90 °C in vacuo for 10 h, while HDI (99 %) and dibutyltin dilaurate (DBTDL) from Merck-Schuchardt (Germany) and HMDI were used as received. The polymerization solvent, N,N-dimethylformamide (DMF, with water content of less than 0.01 %), was purchased from Sigma-Aldrich (Germany) and used as received. The solvent was stored over activated 3–4 Å molecular sieves.
Measurements
Reduced viscosities (η reds, dL g−1) of 0.5 % polymer solution in 1,1,2,2-tetrachloroethane (TChE) were measured in an Ubbelohde viscometer (Poland) at 25 °C.
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were obtained with a Bruker Tensor 27 FTIR spectrometer (Germany) using thin films. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 32 scans per spectrum with a resolution of 4 cm−1.
Elemental analysis was performed with a PerkinElmer CHN 2400 analyzer (USA).
Thermogravimetry (TG) was carried out with a Netzsch STA 449 F1 Jupiter thermal analyzer (Germany) in the range of 40–1000 °C in helium (flow = 20 cm3 min−1), at the heating rate of 10 °C min−1. Sample weights about 10 mg were used. The composition of the gas evolved during the decomposition process was analyzed by a Bruker Tensor 27 FTIR spectrometer (Germany) coupled online to a Netzsch STA instrument by Teflon transfer line with 2 mm diameter heated to 200 °C. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 16 scans per spectrum at 4 cm−1 resolution.
Differential scanning calorimetry (DSC) curves were obtained with a Netzsch 204 calorimeter (Germany) in the range of −100 to 200 °C. The reported transitions were taken from first and second heating scans. The scans were performed at the heating/cooling rate of 10 °C min−1 under nitrogen atmosphere (flow = 30 mL min−1). Sample weights of about 10 mg were used. Glass-transition temperatures (T gs) for the polymer samples were taken as the inflection point on the curves of the heat-capacity changes. Melting temperatures (T ms) were read at endothermic-peak maxima.
Atomic force microscopy (AFM) was carried out on a Nanoscope V (VEECO, USA) microscope, in tapping mode in air. The phase data were recorded simultaneously. Silicon probes that were used (NSG30, NT-MDT, Russia) had a nominal spring constant of 20–100 N m−1. In order to compare the structure of all investigated samples, the imaging parameters were kept constant. Medium tapping technique was used to obtain phase images at 1-μm scan size. The specimens used were the cuttings from crude polymers after one month.
X-ray diffraction (XRD) measurements were performed using a Panalytical Empyrean apparatus (Holland) with a copper tube, nickel filter and focusing mirror. The XRD patterns of the investigated samples were obtained by measuring the number of impulses within a given angle over 4 s. The measurements were taken every 0.01°. The XRD patterns were analyzed by the WAXSFIT computer program [21]. The program resolves a diffraction curve on diffraction peaks and an amorphous halo which allows to estimate the degree of crystallinity. It was calculated as the ratio of the sum of crystalline to the total sum of crystalline and amorphous peaks areas. As crystalline peaks were assumed, those whose full width at half-maximum (FWHM) were contained in the range of 0.5°–1.8°.
The hardness of the polymers was measured by the Shore A/D method on a Zwick 7206/H04 hardness tester (Germany) at 23 °C; and values were taken after 15 s.
Tensile testing was performed on a Zwick/Roell Z010 (Germany) tensile-testing machine according to Polish Standard PN-81/C-89034 (EN ISO Standard 527-1:1996 and 527-2:1996) at the speed of 100 mm min−1 at 23 °C; tensile test pieces 1 mm thick and 6 mm wide (for the section measured) were cut from the pressed sheet.
Press molding was done with a Carver hydraulic press (USA) at 80–140 °C under 10–30 MPa pressure.
The single-lap shear strength of the polymers to copper plate, 100 mm × 25 mm × 1.5 mm, was measured in accordance with Polish Standard PN-EN 1465:2009 by using a Zwick/Roell Z010 (Germany). The adhesive joint, 12.5 mm × 25 mm × 0.2 mm, was prepared by pressing the polymer between the ends of two copper plates at 80–120 °C (prepared according to PN-EN-13887:2005) and then leaving them under a pressure of 30 MPa to cool to room temperature. Next, the plates were fixed by tensile-testing machine clips and underwent tensile testing, the speed of 2 mm/min at 23 °C.
Refractive index was measured at 23 °C by using Conbest Abbe’s Refractometer Type 325 instrument (Poland) according to method A of European Standard EN ISO 489:1999. 1-Bromonaphtalene was applied between the sample film and the prism shield.
The ultraviolet–visible (UV–Vis) spectra of the compression-molded sheets, 1-mm-thick sheets, of the TPUs were determined by a UV-1800 (Shimadzu, Japan) UV spectrophotometer in the range of 300–900 nm, sampling interval 0.5 nm.
Polymer synthesis
RPURs
The RPURs were prepared by solution (DMF, conc. ~20 mass%) polyaddition of the diol OSOE and HMDI or HDI at the NCO/OH molar ratio of 1.05. The reactions were carried out under dry nitrogen for 4 h at 85–90 °C in the presence of a catalytic amount of DBTDL. The polymers precipitated and then washed with methanol were dried at 90 °C in vacuo to constant mass. Yield ~99 %.
TPUs
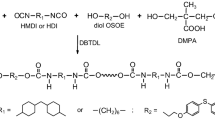
The TPUs, with the soft-segment content of 30, 45 and 60 mol%, were prepared, according to Scheme 1, by a one-step catalyzed melt polyaddition of the diol OSOE, HMDI or HDI and PCD at the NCO/OH molar ratio of 1.05.

Synthesis of TPUs
The general procedure for the synthesis of the TPUs by this method was as follows. The diol OSOE, PCD (0.01 mol together) and diisocyanate (0.0105 mol) were heated with stirring under dry nitrogen to 110 °C in an oil bath. A catalytic amount of DBTDL (about 0.02 g) was added to the formed clear melt and polymerization rapidly began at vigorous stirring. The reaction temperature was gradually raised to 135 °C, and the formed colorless rubber-like product was additionally heated at this temperature for 2 h.
Results and discussion
Polymer characterization
The thermoplastic RPURs, synthesized as models of hard segments in the TPUs, were colorless materials with the similar η red values. Polymer HM showed good solubility in common organic solvents, contrary to polymer H, which was completely soluble only in TChE. The η red values, DSC, TG and XRD data received for these polymers are given in Tables 1–4.
The related TPUs were colorless, transparent or opaque materials. They did not become yellow after a long exposure (6 months) to atmospheric conditions at room temperature in contrast to MDI-based ones. The η red values determined for these polymers, ranging from 0.85 to 4.15 dL g−1, pointed to high-molar masses, especially of the TPUs with 45 and 60 mol% content of soft segments. The HMDI-based TPUs were considerably more soluble in common organic solvents in comparison with the HDI-based ones. All these polymers dissolved at room temperature in TChE, N-methyl-2-pyrrolidone (NMP), N,N-dimethylformamide, tetrahydrofuran and N,N-dimethylacetamide (except for HM-30), but they were insoluble in dimethyl sulfoxide. In the HDI series, polymer H-60 was completely soluble in TChE (at room temperature) and NMP (at elevated temperature), whereas polymer H-45 dissolved only in TChE (at elevated temperature). The remaining TPUs were partially soluble or only swelled.
Table 1 gives designations, η red values and transmittance data for the TPUs.
The chemical structures of all the polymers were confirmed by ATR–FTIR spectroscopy, and in the case of RPURs also by elemental analysis.
FTIR (cm−1)
RPUR from HMDI: 1701 (H-bonded C=O stretching), 1511 (N–H bending) and 3335 (N–H stretching) of the urethane group; 2922 and 2852 (asymmetric and symmetric C–H stretching of CH2); 3059 (C–H stretching) and 1592 and 1489 (C–C stretching) of benzene ring; 822 (C–H bending of p-disubstituted benzene ring); 1218 (asymmetric) and 1046 (symmetric) C–O–C stretching in aliphatic–aromatic ether; 1451 (C–H bending of cyclohexane ring).
RPUR from HDI: 1684 (H-bonded C=O stretching), 1535 (N–H bending) and 3318 (N–H stretching) of the urethane group; 2933 and 2858 (asymmetric and symmetric C–H stretching of CH2); 3059 (C–H stretching) and 1593 and 1492 (C–C stretching) of benzene ring; 829 (C–H bending of p-disubstituted benzene ring); 1241 (asymmetric) and 1070 (symmetric) C–O–C stretching in aliphatic–aromatic ether.
TPUs from HMDI: 1523–1521 (N–H bending) and 3376–3369 (N–H stretching) of the urethane group; 1739–1718 (C=O stretching of the urethane and carbonate groups); 1245–1242 (C–O stretching of the carbonate group and asymmetric C–O–C stretching in aliphatic–aromatic ether); 1051 (symmetric C–O–C stretching in aliphatic–aromatic ether); 792–791 (out-of-plane bending of O–CO–O); 2934–2929 and 2860–2857 (asymmetric and symmetric C–H stretching) and 1460–1457 (asymmetric C–H bending) of CH2; 1593 and 1492 (C–C stretching of benzene ring); 830–822 (C–H bending of p-disubstituted benzene ring).
TPUs from HDI: 1536–1535 (N–H bending) and 3326–3320 (N–H stretching) of the urethane group; 1738–1683 (C=O stretching of the urethane and carbonate groups); 1243–1242 (C–O stretching of the carbonate group and asymmetric C–O–C stretching in aliphatic–aromatic ether); 1057–1054 (symmetric C–O–C stretching in aliphatic–aromatic ether); 792–791 (out-of-plane bending of O–CO–O); 2937–2936 and 2866–2861 (asymmetric and symmetric C–H stretching) and 1464–1458 (asymmetric C–H bending) of CH2; 1593 and 1492 (C–C stretching of benzene ring); 831–822 (C–H bending of p-disubstituted benzene ring).
Elemental analysis
RPUR from HMDI: Calcd for C31H40N2O6S: C, 65.47 %; H, 7.09 %; N, 4.93 %; found: C, 65.25 %; H, 7.23 %; N, 4.93 %.
RPUR from HDI: Calcd for C24H30N2O6S: C, 60.74 %; H, 6.37 %; N, 5.90 %; found: C, 60.42 %; H, 6.46 %; N, 6.43 %.
Figure 1 shows the spectra of the RPURs and TPUs with 30 and 60 mol% PCD content.

FTIR spectra of the RPURs and TPUs containing 30 and 60 mol% PCD
All the FTIR spectra exhibited significant absorptions of the urethane and ether groups, benzene ring and methylene group. In the case of the TPUs, absorption of carbonate group was also observed. Polyurethanes require an analysis of the carbonyl stretching region, which gives some information about their order of hard domains or degree of microphase separation.
The spectrum of the RPUR H showed a sharp band at 1684 cm−1, while that of the RPUR HM exhibited a broader one at 1701 cm−1, characteristic of the H-bonded urethane carbonyl groups [22, 23], in well-ordered “crystalline” structures and disordered “amorphous” ones, respectively. This was in agreement with the results of DSC and XRD analyses.
In the spectra of the HDI-based TPUs, two bands at 1738–1736 cm−1 (more intensive) and 1688–1683 cm−1 were visible, corresponding to the nonbonded carbonate carbonyl groups and H-bonded urethane carbonyl groups in ordered hard-segment domains, respectively [24]. With the growth of the soft segment content, a progressive increase in the intensity of the peak at the higher wavenumber, with a simultaneous decrease in the intensity of the peak at the lower wavenumber, was observed. This was connected with a reduction of ordered hard-segment domains in the polymer. In the case of the HMDI series, all the spectra displayed a band at 1739–1735 cm−1 corresponding to the nonbonded carbonate carbonyl groups, but did not show the band at 1701 cm−1 characteristic of H-bonded urethane carbonyl groups in disordered “amorphous” structures, which was observed for RPUR HM. That can be explained by a considerable phase mixing in these TPUs (see DSC data in Table 2). In the spectrum of polymer HM-30, however, a band at 1718 cm−1 was visible, missing in the spectra of the polymers with lower hard-segment content, which can be assigned to the absorbance of nonbonded urethane carbonyl groups.
Thermal properties
DSC
The numerical data obtained (T g, T m and heat of melting (ΔH) values) for all the TPUs and RPURs after one-month conditioning at room temperature, as well as for PCD, are summarized in Table 2, while the DSC curves of all these polymers are presented in Figs. 2 and 3.

DSC curves of the HMDI-based polymers after one-month conditioning (HM, HM-30, HM-45 and HM-60) and additionally after three-month conditioning (HM-60a) as well as PCD

DSC curves of the HDI-based polymers after one-month conditioning (H, H-30, H-45 and H-60) and additionally after 3-month conditioning (H-60a) as well as PCD
The DSC curve of RPUR H (model of the HDI-based hard segment in the related TPUs) from the first heating scans showed glass transition at 86 °C and one endothermic peak with maximum at 151 °C. High ΔH value (70.5 J g−1) of this endothermic transition pointed to a high degree of ordering of this polymer. In the second heating scan, besides a more distinct glass transition (48 °C), was observed only a small endothermic peak at maximum at 163 °C. The DSC curves of all the related TPUs from the first heating scans displayed, apart from glass transition of the soft segment in the range of −35 to −31 °C, a few endothermic peaks at maxima in the range of 17–174 °C. Comparing the data obtained for pure soft segment (PCD) and TPUs, it can be seen that endothermic peaks with maxima at 17–60 °C are associated with the melting of soft-segment domains, while those at higher temperatures (80–174 °C) are connected with the melting of hard-segment domains. The lower-temperature peaks at 80–87 °C could be ascribed to the melting of the less ordered structures, whereas those at 168–174 ° correspond to the melting of the more ordered structures. With the increase of the PCD content, the ordering in hard-segment domains diminished, but grew in soft-segment domains, though to a lesser extent. Polymer H-30 contained well-ordered hard-segment domains and practically disordered soft-segment domains, whereas polymer H-60 with the highest soft-segment content was characterized by a low ordering of hard-segment and soft-segment domains. All the HDI-based TPUs showed a relatively high degree of microphase separation (little difference between T g values of pure PCD soft segments and the polymer) almost independent of polymer composition. On the DSC curves from the second heating scans, only glass transition, shifted to higher temperatures, was observed. This shift was caused by an increase in the mutual miscibility of the soft and hard segments during the first heating.
The DSC curve of RPUR HM (model of the HMDI-based hard segment in the related TPUs) from the first heating scans displayed glass transition at 72 °C and two broad overlapping endothermic peaks with maxima at 133 and 167 °C. Low ΔH value (8.7 J g−1) of this endothermic transition, in comparison with that of RPUR H, indicated a much lower degree of ordering of polymer HM. In the second heating scan, only glass transition (94 °C) was visible. The DSC curves of related TPUs, both from the first and second heating scans, showed glass transition at higher temperatures than the analogous polymers derived from HDI, dropping with the increase of the soft-segment content. This points to a lower degree of microphase separation of the HMDI-based polymers, just as it was found by other authors [25]. The presence of no endothermic peaks on the curve (HM-45) or only very small ones, i.e., at 40 °C (ΔH 0.8 J g−1) for HM-60 and at 169 °C (ΔH 0.6 J g−1) for HM-30, corresponding to the melting of soft-segment domains and hard-segment domains, respectively, suggests amorphous structures of all the HMDI-based TPUs.
A heterogeneous bulk morphology of the examined TPUs was confirmed by the phase AFM images (with 1 µm scan size) shown in Fig. 4. The dark matrix of lower modulus corresponds to the soft-segment-rich domains and the brighter dispersed domains of higher modulus correspond to the hard segments. The hard-segment-rich domains were features of different size and shape, depending on the kind of diisocyanate component and the stoichiometry between the hard and soft segments. The greatest differences in the appearance of the images can be seen for polymers with the highest content of the hard segments (H-30 and HM-30), which differed the most in the degree of microphase separation and at the same time in the degree of ordering within the hard-segment domains. Thus, the AFM image of HM-30 shows mainly short, rather densely packed and randomly oriented rod-like structures, while in the case of polymer H-30, apart from smaller objects, numerous greater aggregates are observed, with a circular rather than rod-like structure.

AFM phase images of the TPUs
These differences are neutralized for polymers with 60 mol% content of soft segments, showing a similar degree of microphase separation.
Moreover, in both series, the fraction of bright hard domains increased with the increase of hard-segment content, which is consistent with the DSC data.
TG
The decomposition process of all the synthesized polymers as well as PCD was performed in inert atmosphere. Moreover, for the RPURs and TPUs with 30 mol% PCD content, an analysis was carried out of volatile decomposition products by the TG coupled with FTIR spectroscopy (TG–FTIR). The TG data obtained are given in Table 3 and Fig. 5, while the FTIR spectra of volatile decomposition products of polymers are presented in Figs. 6–8.

DTG and TG curves of polymers HM and HM-30

FTIR spectra of volatile products obtained at the maximum rate of mass loss of the thermal decomposition of RPURs H and HM (for the first and second stages)
The polymers were stable up to 262–278 °C taking into account T 1 as a criterion of stability, with somewhat higher values shown by the polymers of the HDI series. These polymers also exhibited higher T 5, T 10, T 50. The results received are consistent with the rule that the more easily formed urethanes are less stable, i.e., they easily dissociate [26, 27], and thus somewhat more reactive HMDI gives polyurethanes with slightly worse thermal stability in comparison with the HDI-based ones. Moreover, it was found that the TPUs showed lower values of all these temperature indicators than the RPURs. It can be caused by a lower degree of ordering of the hard segments. Considering T 1, T 5 and T 10 values received for PCD, it may be assumed that a worse TPUs stability in the early stage of decomposition is also due to the worse stability of the polycarbonate soft segment than that of hard segments. However, with the rise of soft-segment content, these indicators somewhat increase. That points to the fact that the decomposition of polycarbonate with hydroxyl groups ending (PCD) takes a different course than that which is incorporated in the TPU chain. In the later stage of decomposition, the stabilities of the soft segment and the hard segments were similar (almost the same T 50 values: 359 °C for H, 353 °C for HM and 354 °C for PCD).
All the obtained polymers, both the RPURs and TPUs, decomposed in two stages. From the course of the TG curves, it results that over 90 % of the polymer is decomposed at the first stage. The DTG curves showed one large intense peak with maximum at 338–364 °C and a small intense peak with maximum at 396–409 °C (for TPUs) and at 428 °C (for RPUR H) and 449 °C (for RPUR HM). From the data given in Table 3, it follows that the ratio of hard to soft segments has a smaller influence on T 50 than T max, the latter in both series increasing with the rise of soft segment. And so, at the highest soft-segment content, T max is observed to shift to slightly higher temperature.
The analysis of FTIR spectra from the first decomposition stage of RPURs (T max at ~360 °C) presented in Fig. 6, showed that their decomposition was connected with the elimination of carbon dioxide (intensive bands at 2359–2310 cm−1, attributed to asymmetric stretching vibration and at 669 cm−1, associated with the degenerate bending vibration), carbonyl sulfide (very low intensity bands at 2072 and 2047 cm−1, characteristic of asymmetric and symmetric C=O stretching vibration [28–32]) and water (bands at ~4000–3500 cm−1 and ~1800–1300 cm−1 [33]).
With the growth of the temperature, the band coming from carbon dioxide decreased, while the one from carbonyl sulfide increased, reaching a maximum at temperature 415 °C (for H) and 380 °C (for HM) (see Fig. 7). Thus, an intensive evolution of carbonyl sulfide occurred at the end of the first stage of decomposition.

3D plots of FTIR spectra of volatile products obtained during the thermal decomposition of the RPURs H and HM
The analysis of the spectra registered from the start of the decomposition additionally showed that the evolution of carbonyl sulfide began at higher temperatures (~360 °C) than carbon dioxide (~270 °C). This indicates that the decomposition of these polymers started from decomposition of a urethane linkage (generating carbon dioxide) and not a sulfide one (generating carbonyl sulfide).
The FTIR spectra recorded during the second decomposition stage for RPURs (T max at 428 °C (for H) and 449 °C (for HM)) exhibited bands typical of carbon dioxide and water, and in the case of polymer H, band characteristics of carbonyl sulfide were still observed. Moreover, the spectra showed bands pointing to the presence of alkenes (at ~2932 and ~2860 cm−1, characteristic of asymmetric and symmetric C–H stretching vibration of methylene and methyl groups and at ~998 and 916 cm−1, connected with C–H out-of-plane deformation vibration of vinyl group) and carbon monoxide (at ~2186 cm−1, attributed to stretching vibration) as well as aromatic compounds (at 748 cm−1, characteristic of C–H out-of-plane deformation vibration of monosubstituted benzenes) and ethers (at 1260 and 1184 cm−1, associated with C–O stretching vibration) only for polymer HM.
From the comparison of spectra obtained for RPURs and diol OSOE [12] (the presence of water and carbon dioxide), it results that decomposition of these polymers was attached to the dissociation of a urethane linkage to a diisocyanate and diol, according to the following reaction:
The resulting isocyanate may then undergo the reaction of condensation with the formation of carbodiimides and carbon dioxide.
On the other hand, on the basis of the absence of bands characteristic of amines in the spectra of the first stage of decomposition as well as in the second stage, except the band at 1260 cm−1, which may also be ascribed to ethers, one should rule out mechanisms in which primary amine, alkenes and carbon dioxide or secondary amine and carbon dioxide are formed [27, 34].
In the FTIR spectra of the volatile products of TPU decomposition (see Fig. 8) from the first stage (T max at 338 °C for HM-30 and 339 °C for H-30), just as in the case of RPURs, one could see absorption bands typical of carbon dioxide and water. In these spectra, no band characteristics of carbonyl sulfide vibrations were detected, because the start of its formation in the RPURs occurred in higher temperature, i.e., ~360 °C. Moreover, all the spectra showed the bands indicating the formation of carbon monoxide (at 2179 and 2184 cm−1 associated with stretching vibration), alcohols (at ~1049 cm−1, associated with C–OH stretching vibration) and ethers (at ~1144 cm−1 attributed to C–O stretching vibration of ether group). The bands pointing to the presence of aliphatic unsaturated compounds (at 2938 and 2864 cm−1, characteristic of asymmetric and symmetric C–H stretching vibration of methylene and methyl groups as well as at ~3085 cm−1, connected with C–H stretching vibration and at ~998 and 916 cm−1—with C–H out-of-plane deformation vibration of vinyl group) were also observed. In the spectra obtained during the second decomposition stage (T max at 401 °C for HM-30 and 402 °C for H-30), there can still be seen bands typical of carbon dioxide, water, carbon monoxide, aliphatic unsaturated compounds and alcohols (probably 5-hexen-1-ol [32]), and there appeared bands pointing to the presence of carbonyl sulfide (bands at 2073 and 2047 cm−1, characteristic of asymmetric and symmetric C=O stretching vibration), aldehydes (at ~2730 cm−1, connected with C–H stretching vibration and at ~1757 cm−1—with C=O stretching vibration of aldehyde group) and aliphatic–aromatic ethers (at ~1258 and 1177 cm−1, associated with C–O stretching vibration of ether group). Taking into consideration products originating from the decomposition of the hard-segment-type polymers, it can be concluded that aliphatic ethers, aldehydes and unsaturated alcohols were the products of the decomposition of the PCD soft segments. Obviously, carbon dioxide is a product of the breakdown of both the hard and soft segments.

FTIR spectra of volatile products obtained at the maximum rate of mass loss of the thermal decomposition of TPUs HM-30 and H-30 (for the first and second stages)
Transparency
In order to obtain transparent elastomers, one should normally use relatively high molar mass oligomer diols as soft segments. On the other hand, such oligomer diols show increased crystallization tendency and thus result in opacity [18]. From the literature and our own research, it follows that this tendency depends on soft-segment content and increases with the growth of its content. DSC studies additionally carried out after three-month conditioning of the polymers at room temperature showed that only in the case of polymer HM-60 were there significant differences in the course of DSC curves. Figure 2 displays curves obtained for polymer HM-60, while Fig. 3—for comparison for H-60. Taking this into account, transparency tests were conducted after 3 months and the results can be found in Table 1 and Fig. 9. In the case of polymers of HMDI series, in which hard-segment domains are amorphous (the lack of distinct endothermic peaks on the DSC curves), transparency is determined by crystallization of soft segments, and thus polymer HM-60 is non-transparent and the remaining two show good transparency. It is different in the case of polymers of the HDI series, where the non-transparent polymer turned to be H-30, showing the highest degree of hard-segment ordering, and a relatively good transparency was exhibited by polymer H-45.

UV–Vis spectra of selected TPUs
XRD analysis
The XRD analysis was done for the polymers, whose DSC curves exhibited endothermic peaks, i.e., all of the HDI series and polymer HM-60. The received XRD patterns, presented in Fig. 10, indicated amorphous structures of polymers H-45 and H-60, and partially ordered ones of the remaining polymers (H, H-30 and HM-60). The analysis of the XRD patterns of these latter polymers by the WAXSFIT program showed that they had partially crystalline structures, and at the same time, the RPUR had a much higher degree of crystallinity than the segmented polymers. Taking into account the DSC data, it could be stated that polymer H-30 contained partially crystalline hard-segment domains, whereas polymer HM-60 partially crystalline soft-segment domains. The results of the studies conducted are given in Table 4 and Fig. 11.

XRD patterns of the polymers based on HDI and TPU HM-60

XRD curves (points) of a RPUR H, b TPU H-30 and c TPU HM-60 resolved into crystalline and amorphous peaks (solid lines)
Refractive index
The measurements of refractive index were made for TPUs which were not visibly opaque, i.e., H-45, H-60, HM-30 and HM-45 and their analogs based on BD. In both series of the TPUs obtained from diol OSOE, the increase of hard-segment content caused the rise of refractive index. While comparing polymers with the same content of the hard segments, i.e., H-45 and HM-45, the former showed a higher value of this parameter. For the TPUs derived from BD, refractive index practically did not depend on the polymer composition, and in each case, the values were lower than those obtained for the TPUs with sulfur atoms.
Mechanical properties
The Shore A/D hardness and tensile properties were determined for all the TPUs after three-month conditioning at room temperature, and the numerical data are shown in Table 5.
The polymers based on “bulky” diisocyanate HMDI showed generally higher hardness, both by using hardness tester of A and D types, in comparison with the analogous TPUs derived from HDI. In the case of these polymers, an increase of hardness was observed with an increment of the soft segment content. Polymer HM-60, possessing a partially crystalline structure, connected with soft-segment crystallization (see Fig. 2), exhibited very high hardness (90°Sh A). In the HDI series TPUs, which were characterized by some degree of ordering, both within hard-segment domains and soft-segment domains, this correlation was not observed.
The obtained TPUs exhibited the modulus of elasticity in the range of 40.8–72.1 MPa, with generally higher values shown by those of the HMDI series. In these series, as the soft-segment content increased, so did the modulus of elasticity, as in the case of hardness.
All the TPUs possessed good tensile strength, ranging from 33.0 to 38.7 MPa. Their elongation at break was contained in the range of 280–470 % (HMDI series) and 300–600 % (HDI series) and in both series increased when the soft-segment content increased.
Adhesive properties
To study the lap shear strengths to copper, we chose the TPUs with the highest hard-segment content (the highest sulfur content), i.e., H-30 and HM-30 and the analogous TPUs based on BD as a chain extender. The obtained results are presented in Fig. 12. They reveal that both polymers HM-30 and H-30 showed over 2-times higher adhesive strength than those from BD (5.8 and 4.4 vs. 2.6 and 2.0 MPa). In the case of both types of polymers, higher lap shear strengths to copper (about 1.3-times) were found for polymers based on HMDI.

Lap-shear strength of selected TPUs and their analogs based on BD
Conclusions
New high molar mass TPUs containing diphenyl sulfide units were synthesized by a one-step catalyzed melt polyaddition of 2,2′-[sulfanediylbis(benzene-1,4-diyloxy)]diethanol (nonconventional chain extender), HMDI or HDI and 30, 45 and 60 mol% aliphatic polycarbonate diol (PCD) of M n = 2000 g mol−1. A DSC study showed that the TPUs of HDI series were characterized by T gs almost independent of the PCD soft-segment content (~−33 °C) and lower than those of the HMDI series (−20 to 1 °C). These investigations also showed that a relatively high degree of microphase separation was exhibited by all the HDI-based TPUs, while in the case of the HMDI-based ones, it grew with the growth of the soft-segment content. This two-phase morphology was supported by AFM. Taking into account the course of DSC curves, it can be said that the TPUs obtained from HDI (with the exception of H-60) showed a higher tendency to forming ordered structures both within hard-segment and soft-segment domains. The crystalline phase was found only for polymers H-30 and HM-60, connected with the hard-segment and soft-segment crystallization, respectively, and these polymers were not transparent. A relatively good transparency was exhibited by TPUs HM-30, HM-45 and H-45. The study conducted for selected TPUs showed their enhanced refractive index and copper adhesion in comparison with the analogous TPUs based on BD. The TPUs were stable up to 262–272 °C, taking into account the T 1, with somewhat higher values shown by those based on HDI. They decomposed in a two-stage process, and the main decomposition (~90 %), both the hard and soft segments, occurred at the first stage. The basic volatile products of the hard-segment decomposition were carbon dioxide, water and carbonyl sulfide, while aliphatic ethers, aldehydes and unsaturated alcohols as well as carbon dioxide originated from the soft-segment decomposition. The analysis of the volatile products showed that the decomposition of the hard segments started from the decomposition of a urethane linkage and not a sulfide one and was connected with its dissociation to a diisocyanate and diol. The synthesized TPUs, with hardness in the range of 80–90°Sh A, possessed a good tensile strength (33.0–38.7 MPa), similar to that of commercial biodurable medical grade TPU (PCD/HMDI/BD), i.e., ChronoFlex® AL 80A (37.9 MPa) [35]. The TPUs obtained by us, both transparent and non-transparent, can be attractive in many medical appliances.
References
Ulrich H. Polyurethanes. In: Mark HF, editor. Encyclopedia of polymers science and technology, vol. 4. New Jersey: Wiley; 2003. p. 26–72.
Sonnenschein MF. Polyurethanes: science, technology, markets, and trends. New Jersey: Wiley; 2014.
Zdrahala RJ. Small caliber vascular grafts. 2. Polyurethanes revisited. J Biomater Appl. 1996;11:37–61.
Resiak I, Rokicki G. Modified polyurethanes for biomedical applications. Polim Wars. 2000;45:592–602.
Hasirci N, Aksoy EA. Synthesis and modifications of polyurethanes for biomedical purposes. High Perform Polym. 2007;19:621–37.
Kultys A, Podkoscielny W, Pikus S. Polyurethanes containing sulfur. I. New thermoplastic polyurethanes with benzophenone unit in their structure. J Polym Sci A Polym Chem. 1999;37:4140–50.
Kultys A, Pikus S. Polyurethanes containing sulfur. III. New thermoplastic HDI-based segmented polyurethanes with diphenylmethane unit in their structure. J Polym Sci A Polym Chem. 2001;39:1733–42.
Rogulska M, Kultys A, Podkoscielny W. Studies on thermoplastic polyurethanes based on new diphenylethane-derivative diols. II. Synthesis and characterization of segmented polyurethanes from HDI and MDI. Eur Polym J. 2007;43:1402–14.
Kultys A, Rogulska M, Pikus S, Skrzypiec K. The synthesis and characterization of new thermoplastic poly(carbonate-urethane) elastomers derived from HDI and aliphatic-aromatic chain extenders. Eur Polym J. 2009;45:2629–43.
Kultys A, Rogulska M, Gluchowska H. The effect of soft-segment structure on the properties of novel thermoplastic polyurethane elastomers based on an unconventional chain extender. Polym Int. 2011;60:652–9.
Kultys A, Rogulska M, Pikus S. New thermoplastic segmented polyurethanes with hard segments derived from 4,4′-diphenylmethane diisocyanate and methylenebis(1,4-phenylenemethylenethio)dialcanols. J Appl Polym Sci. 2012;123:331–46.
Rogulska M, Kultys A, Lubczak J. New thermoplastic polyurethane elastomers based on aliphatic-aromatic chain extenders with different content of sulfur atoms. J Therm Anal Calorim. 2015;121:397–410.
Kultys A, Rogulska M, Pikus S. The synthesis and characterization of new thermoplastic poly(thiourethane-urethane)s. J Polym Sci A Polym Chem. 2008;46:1770–82.
Rogulska M, Kultys A, Olszewska E. New thermoplastic poly(thiourethane-urethane) elastomers based on hexane-1,6-diyl diisocyanate (HDI). J Therm Anal Calorim. 2013;114:903–16.
Kultys A, Puszka A. Transparent poly(thiourethane-urethane)s based on dithiol chain extender. Synthesis and characterization. J Therm Anal Calorim. 2014;117:1427–39.
Kultys A, Puszka A. New thermoplastic polyurethane elastomers based on sulfur-containing chain extenders. Pol J Chem Technol. 2013;15:1–6.
Lee DK, Tsai HB, Tsai RS, Chen PH. Preparation and properties of transparent thermoplastic segmented polyurethanes derived from different polyols. Polym Eng Sci. 2007;47:695–701.
Hepburn C. Trends in polyurethane elastomer technology. Iran J Polym Sci Technol. 1992;1(2):84–110.
Chen PH, Yang YF, Lee DK, et al. Synthesis and properties of transparent thermoplastic segmented polyurethanes. Adv Polym Technol. 2007;26(1):33–40.
Penczek S, Frisch KC, Szczepaniak B, Rudnik E. Synthesis and properties of liquid crystalline polyurethanes. J Polym Sci A Polym Chem. 1993;31:1211–20.
Rabiej M, Rabiej S. Analysis of X-ray diffraction pattern of polymers by means of WAXSFIT computer program (in Polish). Poland: ATM; 2006.
Eceiza A, Martin MD, de la Caba K, Kortaberria G, Gabilondo N, Corcuera MA, Mondragon I. Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: mechanical and thermal properties. Polym Eng Sci. 2008;48:297–306.
Rueda-Larraz L, Fernandez B, Tercjak A, Ribes A, Mondragon I, Eceiza A. Synthesis and microstructure–mechanical property relationships of segmented polyurethanes based on a PCL–PTHF–PCL block copolymer as soft segment. Eur Polym J. 2009;45:2096–109.
Hernandez R, Weksler J, Padsalgikar A, Runt J. In vitro oxidation of high polydimethylsiloxane content biomedical polyurethanes: correlation with the microstructure. J Biomed Mater Res. 2008;87A:546–56.
Tang YW, Labow RS, Santerre JP. Enzyme-induced biodegradation of polycarbonate-polyurethanes: dependence on hard-segment chemistry. J Biomed Mater Res. 2001;57:597–611.
Wirpsza Z. Polyurethanes: chemistry, technology and applications. New York: Ellis Horwood; 1993.
Chattopadhyay DK, Webster DC. Thermal stability and flame retardancy of polyurethanes. Prog Polym Sci. 2009;34:1068–133.
Dunn JG, Chamberlain AC, Fisher NG, Avraamides J. The influence of activated carbon on the thermal decomposition of sodium ethyl xanthate. J Therm Anal Calorim. 1997;49:1399–408.
Fisher NG, Dunn JG. Analysis of a complex gaseous mixture by TG–MS and TG–FTIR. J Therm Anal Calorim. 1999;56:43–9.
Madarász J, Pokol G. Comparative evolved gas analyses on thermal degradation of thiourea by coupled TG–FTIR and TG/DTA-MS instruments. J Therm Anal Calorim. 2007;88:329–36.
Otto K, Bombicz P, Madarász J, Oja Acik I, Krunks M, Pokol G. Structure and evolved gas analyses (TG/DTA-MS and TG–FTIR) of mer-trichlorotris(thiourea)-indium(III), a precursor for indium sulfide thin films. J Therm Anal Calorim. 2011;105:83–91.
Ahamad T, Alshehri SM. Thermal degradation and evolved gas analysis of thiourea-formaldehyde resin (TFR) during pyrolysis and combustion. J Therm Anal Calorim. 2012;109:1039–47.
The website http://webbook.nist.gov/chemistry/.
Simon J, Barla F, Kelemen-Haller A, Farkas F, Kraxner M. Thermal stability of polyurethanes. Chromatographia. 1988;25:99–106.
The website http://www.matweb.com.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rogulska, M., Kultys, A. Aliphatic polycarbonate-based thermoplastic polyurethane elastomers containing diphenyl sulfide units. J Therm Anal Calorim 126, 225–243 (2016). https://doi.org/10.1007/s10973-016-5420-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5420-z