
Abstract
The reactivity of a system comprising commercial epoxy resin Epidian® 6 (E6) and triethylenetetramine (Z-1) hardener was studied. The system was modified with two low-molecular siloxane modifiers: 1,3-bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane (DS1) and 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane (DS2). The modifiers replaced 10 mass% of E6 or Z-1, respectively. The systems E6/DS2 and DS1/Z-1 were also studied. Enthalpy of curing was determined for all systems in isothermal conditions and dynamically, i.e., in the range of 0–250 °C with linearly increasing temperature at the rate of 2, 5, 10, and 20 °C min−1. The activation energy of curing was determined using isoconversional methods by Friedman and Kissinger–Akahira–Sunose. The epoxy systems containing siloxane modifiers had generally lower activation energies than the reference E6/Z-1 system. The time to gelation measurements were taken using the temperature-modulated differential scanning calorimetry.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epoxy resins are today one of the most important group of polymeric materials [1–8].Among their advantages are the relatively high thermal stability and good whether resistance as well as resistance against many chemicals. Polar character of the resins determines their very good adhesion to materials such as metals, glass, concrete, and sufficiently polar polymers. Epoxy composites reinforced with glass or carbon fibers have excellent mechanical properties and are widely used in aerospace industry.
The properties of materials based on epoxy resins can be widely changed by selecting appropriate curing agent or by introducing extra modifiers or active diluents replacing a part of resin or curing agent. One of the most commonly used group of curing agents for liquid epoxy resins are aliphatic [9–14], or aromatic [15–18] amines or imidazole derivatives [19, 20]. Recently, among frequently studied modifiers of epoxy polymers are silicone containing compounds, including various siloxanes or silsesquioxanes [21–30]. The effect of two low-molecular siloxanes: 1,3-Bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane and 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane on performance properties of liquid bisphenol A epoxy resin cured with triethylenetetramine were reported in our previous paper [25].
A key factor when developing new thermoset materials is to assure possibly high conversion of components. The appropriate tool to study the problem is the thermal analysis, in particular the conventional or temperature-modulated differential scanning calorimetry (DSC). The DSC study can be carried out in a dynamic [9, 31–38] or isothermal mode [22, 39–41].
There exist several generally phenomenological models providing quantitative information on the activation energy and kinetics of curing. Among them, the most popular are the models of Friedman, Kissinger–Akahira–Sunose (KAS) or Ozawa–Flynn–Wall. The equations are provided by the software systems of modern thermal analysis instruments. More methods of reactivity studies were developed while interpreting the results of dynamic torsional vibration [20] and spectroscopic measurements [42–44].
In this paper, we test different methods of evaluating activation energy of curing of few epoxy systems as available from dynamic and isothermal DSC. The same experiments were also applied to determine kinetic coefficients of curing processes using several phenomenological models.
Experimental
Materials
A commercial grade diglycidyl ether of bisphenol A: Epidian® 6 (E6, epoxy number 0.538 equiv./100 g or equivalent epoxy mass 185.9 g mol−1) and triethylenetetramine (Z-1) were products of Organika-Sarzyna Chemical Plants (Poland). 1,3-Bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane (DS1, epoxy number 0.534 equiv./100 g or equivalent epoxy mass 187.3 g mol−1) was prepared as described in our previous paper [25]. 1,3-Bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane (DS2) was a product of Gelest Inc. (USA).
Samples
The samples were prepared by weighing the components of each system studied (ca. 2 g in total), careful homogenizing the liquid mixture, and placing 20 mg of it in an aluminum crucible. The liquids were transparent, except for composition DS1/Z-1 which was slightly opaque.
Reactivity measurements by DSC
Reactivity of the system E6/Z-1 consisting of a liquid epoxy resin E6 and aliphatic amine: triethylenetetramine (Z-1) was measured. Furthermore, epoxy systems were studied where 10 % by mass of each component, E6 and Z-1, was replaced by reactive modifiers: 1,3-bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane (DS1) and 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane (DS2), respectively. Finally, the systems consisting of the epoxy siloxane monomer were cured with triethylenetetramine (DS1/Z-1) and Epidian 6 with amino-siloxane component (E6/DS2). The formulas of the components are presented in Scheme 1.

The components of epoxy systems
Equimolar ratios of epoxy groups to amino hydrogens were used in all systems. Both epoxy monomers were considered bifunctional, whereas amino monomers Z-1 and DS2 were hexa- and tetrafunctional, respectively.
The DSC measurements were taken on a Mettler Toledo DSC1 apparatus equipped with Stare software in nitrogen flowing at the rate of 60 mL min−1. The dynamic experiments were made in temperature range of 0–250 °C with the heating rates of 2, 5, 10, and 20 °C min−1. The isothermal measurements were taken in the range of 40–100 °C at 10° intervals. The lowest temperature in the range (40 °C) was selected to be somewhat higher than room temperature at which the curing processes actually started to proceed. The only exception was the system DS1/Z-1 with 60 °C the lowest constant curing temperature, as below it the system reacted too slowly.
Curing experiments were also made using temperature-modulated DSC. They were used to determine the TT (time–temperature) diagram. The measurements were taken with ten degrees intervals in the range 40–90 °C for all compositions yielding glassy material at room temperature, i.e., for the systems E6/Z-1, E6/DS2, E6/10 %DS1/Z-1, and E6/10 %DS2/Z-1. Only for the system E6/DS2, the highest temperature employed was 70 °C, since the runs carried out at higher temperature did not allowed to determine the time to glass transition. The modulation conditions were as follows: temperature amplitude 0.5 °C, random pulse duration 15–30 s average heating rate 1 °C min−1. The sample preparation method as well as their size was the same in both isothermal and dynamic measurements.
Temperature of glass transition
The glass transition temperature was determined by DSC as the temperature of the fastest jump in the heat flow for the samples both cured isothermally or dynamically. The crucibles with cured material were heated twice in the range 0–250 °C at the rate 10 °C min−1 with nitrogen flow rate 60 mL min−1. The exception was the system DS1/Z-1 yielding an elastic body at room temperature. For this system, the starting temperature was –120 °C.
Results and discussion
Dynamic reactivity
The curing reaction in epoxy system is an exothermic one. Hence, a convenient method of following the reaction is to record the thermal effects accompanying the process by a DSC machine. The dynamic measurements, i.e., the measurements at constant heating rate, provide the temperature range at which the reaction takes place and the temperature of exothermic peak of curing. From the recorded curves, one can calculate the reaction rate and the enthalpy (heat) of the process. The isothermal measurements, on the other hand, provide information on to what extent and at what time the reaction mixture reaches its final conversion.
The simplified chemistry of the epoxy curing by amine hardener is presented on Scheme 2. It includes the reaction of amino group with epoxy ring to produce a hydroxyl group from the epoxy oxygen (I). The product contains secondary amino groups capable of further reaction with another epoxy groups (II). The second reaction step is slower than the first one and produces branching and, eventually, gel formation.

The simplified chemistry of the epoxy curing by amine hardener
The enthalpy and temperature of exothermic curing peak recorded in the dynamic mode, read out from respective curves, are presented in Table 1.
A typical curve of curing the epoxy system is shown in Fig. 1.

DSC curves of curing of epoxy composition E6/Z-1 at heating rates of 2, 5, 10, and 20 °C min−1 in nitrogen atmosphere
As expected, the rate of heating did practically not change the enthalpy of curing for the neat system E6/Z-1. It remained in the range of −95.6 to −98.3 kJ mol−1. The temperature of exothermic maximum, however, did change from 74.6 °C at the lowest heating rate to 114.0 °C at the rate of 20 °C min−1 as shown in Table 2.
The system where the low-molecular siloxane with amino groups was used instead of triethylenetetramine had less negative enthalpy by ca. 12 kJ mol−1 regardless of the heating rate (Table 1). The reason might be the limited miscibility of the components and hence smaller accessibility of functional groups for reaction. The temperatures of exothermic peak for that system were higher than that for E6/Z-1 by 28.5–43.8 °C and increased with growing heating rate.
For the system E6/DS2, the enthalpies of exothermic curing process were by over 11 kJ mol−1 more negative than those for the reference E6/Z-1 one. It can be explained by the presence in the hardener of only primary amino groups that are more reactive than the secondary groups in Z-1.
One should also bear in mind that the hexafunctional Z-1 hardener produced denser network, and hence, the access of all amino protons to epoxy groups might have been sterically hindered. The peak temperatures for E6/DS2 were by ca. 10 °C higher compared to those for E6/Z-1.
Much smaller changes in both enthalpy of curing and position of exothermic maximum temperature were recorded when 10 mass% of either epoxy resin E6 or Z-1 hardener were replaced with the low-molecular siloxane modifiers. Again, an increase in absolute enthalpy value by several kJ mol−1 was observed for the system with 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane hardener as compared to the system with Z-1 alone (Table 2).
Isothermal experiments
The key element of reactivity studies by DSC are the measurements carried out at constant temperature. The total curing time measured as the time elapsed until any heat flow was noted in the curves is presented in Table 3. The enthalpies measured in isothermal experiments at different temperatures are shown in Table 4.
The values of curing enthalpies were used to evaluate the conversion degree of functional groups in the epoxy compositions. The conversion for each of them was calculated as the ratio of the enthalpy measured at a given temperature at the isothermal run (Table 4) to the average value measured in the dynamic analysis (Table 1). The results are shown in Fig. 2. As it is well known, the higher the conversion of bisphenol A-based epoxy resins , the higher the curing temperature [45]. Hence, for the system DS1/Z-1, not derived from bisphenol A, the enthalpies of curing were more or less the same, ranging from −81.9 to −85.8 kJ mol−1, independently of curing temperature. The time of curing was significantly decreased with increasing temperature (Table 3).

The effect of curing temperature on the conversion degree of epoxy compositions
The lowest value of enthalpy, equal to −74.2 kJ mol−1, was recorded for the reference system E6/Z-1 cured at 40 °C. This value corresponded to 76 % conversion assuming full conversion in the dynamic experiments. The curing heat increased with each 10 °C temperature rise by 2.5–7.1 kJ mol−1, to reach the highest enthalpy of −97.0 kJ mol−1 at curing temperature of 100 °C. This enthalpy corresponded to 99.4 % conversion, relative to that recorded in dynamic measurements. The time of curing became shorter with increasing temperature. At 100 °C, it was just 25 min, i.e., more than ten times shorter compared to that at 40 °C (300 min, Table 3).
Interesting behavior was observed for the system E6/DS2. The curing enthalpy at temperature 40 °C was −97.7 kJ mol−1, corresponding to conversion of 88.7 %, whereas almost full conversion (99 %) was attained already at 80 °C. This data illustrate the higher flexibility of the system with siloxane-based cross-linker.
The DSC analysis of the mixed systems with 10 % of either epoxy resin or aliphatic amine cross-linker replaced by their siloxane counterparts (DS1 or DS2) also provided interesting results. Both modifiers yielded a higher heat of curing. For the system E6/10 %DS2/Z-1, it could be attributed to the flexibility and relatively higher reacting heat of curing with cross-linker DS2. On the other hand, no effect of DS1 presence in the system E6/10 %DS1/Z-1 was observed compared to E6/Z-1 one, despite the significantly lower curing heat measured for the system DS1/Z-1. The reason might be that tri-component system, unlike the latter one (DS1/Z-1), was perfectly homogeneous, just like the reference E6/Z-1 one. The presence of both modifiers, however, reduced the curing time.
Glass transition diagram
An important part of reactivity studies in epoxy systems provide the measurements of the time of reaction required for the system to reach glass transition. The relationship between temperature versus time to vitrification is an important part of TTT (time–temperature–transformation) diagrams devised for epoxy polymers by Gillham [46, 47]. The glass transition, i.e., transformation of the system from liquid to glassy state is accompanied by a jump in the specific heat of the resin. The specific heat can conveniently be measured by using a temperature-modulated differential scanning calorimeter. The time to reach glass transition can just be read out from the plot of specific heat versus time. An isothermal curing experiment with temperature modulation also provides a distinction between reversible and irreversible heat flow. In the curing experiments carried out in this work, using TOPEM temperature modulation system, the reversible heat flow, however, was found null, as expected. All heat flow was the irreversible one.
The relationships between temperature of isothermal curing and time to vitrification are shown in Fig. 3 for systems E6/Z-1, E6/DS2, and the tri-component ones. At all temperature levels, the shortest time to reach the cross-linking-induced vitrification was recorded for the reference E6/Z-1 system. With DS2 cross-linker, 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane, the time to vitrification was longer by 70 min at the lowest curing temperature of 40 °C and by 40 min at the highest temperature of 70 °C, for which the glass transition for the system was achievable. Above this temperature, the system E6/DS2 did not vitrify. The systems with 10 % of siloxane modifiers present had also the vitrification times moved toward higher values. Interestingly, the higher the curing temperature, the shorter the times to cross-linking-induced vitrification of siloxane modified epoxy systems.

Glass transition diagram for epoxy systems studied in this work
Kinetics of curing
Kinetic analysis of a complex chemical process is about finding a relationship between the rate of the process, conversion, and temperature [48]. More general, a solution is to evaluate three kinetic parameters in the Arrhenius equation:
the activation energy, E a, pre-exponential factor, A, and the kinetic model f(α) with α, the reaction degree (fractional conversion), that defines the rate coefficient k(T):
R is the gas constant (8.314 J mol−1 K−1). For the DSC experiments, the reaction degree α is assumed as:
where H represents the DSC heat flow, t is the reaction time, and ΔH is the overall reaction exothermal value.
Several methods of evaluation the kinetics of curing or thermal decomposition [49] were devised [50–55] assuming the kinetic model function f(α) does not change with temperature.
Isoconversional methods
Two isoconversional methods by Friedman [56] and KAS [57] were employed to evaluate kinetic parameters of curing under non-isothermal conditions. These methods may be used to determine and to monitor changes in the activation energy during the curing process, without assuming a reaction model.
The Friedman isoconversion method is based on the following equation:
In order to find the apparent activation energy value (E a) for a given degree of conversion (α), one should take a series of measurements at different heating rates β. Then, for a fixed degree of conversion (α = const), straight lines are obtained in the plot of \( \ln \left( {\frac{{{\text{d}}\alpha }}{{{\text{d}}t}}} \right) \) vs. \( 1/T \) for which the slope is defined as \( n = - \frac{{E_{\text{a}} }}{R} \).
On the other hand, the KAS isoconversion method is based on the equation:
Then, with the same data, straight lines are obtained in the plot of \( \ln \left( {\frac{\beta }{{T^{2} }}} \right) \) vs. \( 1/T \) for which the slope is defined as \( m = - \frac{{E_{\text{a}} }}{R} \).
The values of activation energies calculated for different conversion using the Friedman and KAS methods are shown in Fig. 4.

The relationship between activation energy and the fractional conversion for epoxy compositions based on Friedman method (a) and Kissinger–Akahira–Sunose method (b)
From the analysis of isoconversional plots, it follows that at the beginning of reaction (α < 0.4), the reduction trend of E a with the increase in conversion is observed. This phenomenon is associated with the decrease in viscosity as the temperature increases. As the viscosity drops down, the energy barrier for the diffusion in the system, required for ensuring effective collision of reactive groups, reduces. Simultaneously, the increase in molecular mass of reacting species renders the viscosity higher [58]. Above α > 0.4, the apparent activation energy becomes stabilized. This phenomenon might be caused by a balance of the diffusion control and autocatalytic process [58].
The neat composition E6/Z-1 and those with 10 mass% of siloxane components reveal similar E a values in the range of 53–56 kJ mol−1. However, the systems DS1/Z-1 and E6/DS2 show lower apparent activation energy in the range of 48–51 kJ mol−1. This can be attributed to the fact that the viscosity of the reactants is lower in the systems containing mainly siloxanes, which enhances the mobility of reactive groups. The apparent activation energies obtained from both Friedman and KAS method assume very similar values.
Nonlinear regression kinetics
The phenomenological rate equation function f(α) reflecting the complex mechanism of curing process can be fitted to experimental data by using methods of nonlinear regression. The types of functions commonly used for that purpose are listed in Table 5. The iterative procedures were used to estimate the kinetic triplet. The calculations were made using Netzsch Thermokinetics 3.1 software. The quality of fitting was assessed using the correlation coefficient and F-test parameter with 95 % confidence level.
By using the values of E a obtained from isoconversional analysis, the rate functions were fitted to the experimental data. The results from the three best functions are listed in Table 6. The criterion was the highest value of correlation coefficient. An illustration of fitting quality is shown in Fig. 5 for E6/Z1 sample.

An example of fitting the model rate function (line) to experimental data (points) for the epoxy composition E6/Z-1 at different heating rates
The best fit of experimental data to a phenomenological model with high correlation coefficient of 0.999 was obtained for the one-step autocatalytic curing reaction model C n. The autocatalytic character of the reaction is well known as the epoxy groups reacting with amino protons produce hydroxyl groups also reacting with epoxy rings. In the fitting procedures, the highest apparent activation energy of 54.5 kJ mol−1 was obtained for the neat system E6/Z-1. The best fitting for the DS1/Z1 system was obtained for Prout–Tompkins’ model (Bna). Moreover, this system revealed the lowest E a value of 47.5 kJ mol−1. The siloxane monomers had lower activation energies, despite the time of curing in both dynamic and isothermal runs. The longer curing time is related to the smaller values of the pre-exponential factor in the Arrhenius equation.
Glass transition temperature
The samples of epoxy systems cured in either dynamic or isothermal conditions were further analyzed by DSC in order to evaluate their glass transition temperature. As it is well known, the glass transition temperature of epoxy resins strongly depends on curing conditions. The presence of additional monomers and the structure of curing agents may also affect the glass transition temperature.
Dynamic measurements
The glass transition temperatures measured in the second (II) and third (III) heating run of the samples cured at different heating rates are shown in Table 7.
Only the signals characteristic for glass transition were observed on all curves recorded in the II and III heating run. No exothermic effects were observed that could have suggested incomplete curing of the epoxy compositions. Hence, all reaction mixtures reached the conversion attainable at the respective curing condition.
The highest values of the thus determined glass transition temperatures were noted for the systems cured at the heating rate of 2 °C min−1. The slowest heating rate at the curing produced the best cured samples with apparently the least disorder resulting from diffusion control of the process. Still higher glass transition temperatures measured at the third heating runs in the range of 0–250 °C seem to suggest that even at the second run, some cross-linking reaction took place, but too small to show up on the respective curve.
The glass transition temperatures for cured E6/Z-1 polymers in the second heating run was 109 °C through 121 °C, depending on heating rate in the first run. For the polymers prepared with 10 % of siloxane modifiers DS1 and DS2, the values of T g were smaller by 13–25 °C than those for unmodified composition. Still lower were these values in the third heating run. In the case of DS1, a monomer of epoxy content and reactivity similar to those of E6 resin, the obvious reason was the higher flexibility of dimethylsiloxane core of the monomer, since cross-linking degree should have been almost identical. In the case of DS2, the cross-linking density of the system E6/10 %DS2/Z-1 could have been smaller, as a part of hexafunctional monomer Z-1 was replaced with equivalent amount of tetrafunctional amine DS2. On the other hand, the linear part of 1,3-bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane is longer than that of 1,3-bis(aminopropyl)-1,1,3,3-tetramethyl-disiloxane. Hence, the effect of the reduced functionality did not exceed the effect of chain length in the two siloxane modifiers.
The smallest changes in T g values caused by the heating rate variations were observed for the neat resin E6 cured with DS2 monomer alone. Independently of heating conditions, the values were in the range of 76–80 °C. Subsequent heating of the cured samples in temperature range of 0–250 °C increased T g value by 2–3 °C. One should notice, however, that the glass transition temperatures were 76–80 °C only, i.e., lower by up to 40 °C than those for E6/Z-1 system. This substantial decrease in T g value cannot be explained by incomplete reaction since the curing heats for the system E6/DS2 (109.7–110.5 kJ mol−1) were even higher than those for the reference E6/Z-1 system (95.6–98.9 kJ mol−1). Also the differences in T g values measured during second and third heating run were very small thus confirming the high conversion in the first one. The marked differences can be explained only in terms of the smaller cross-linking density of the system E6/DS2, compared to that of E6/Z-1 and larger proportion of flexible siloxane monomer. Indeed, triethylenetetramine (Z-1) has 24.4 atomic mass units per functionality, while DS2 monomer has 61.1 units, not to mention the distances between nitrogen atoms in the two cross-linking agents.
Very small differences in T g values caused by variation in heating rate at curing were observed for the compositions DS1/Z-1. The values were in the range of −28 to −26 °C. The subsequent heating cycle in the range of 0–250 °C resulted in only a minute increase in T g by at most 3 °C proving the full conversion in the first heating run.
Isothermal measurements
The compositions prepared by isothermal curing were also studied by DSC. The resulting T g values measured in second and third heating runs are presented in Table 8.
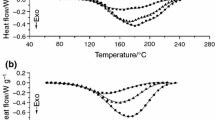
Typical DSC curves for E6/10 %DS1/Z-1 samples cured isothermally at different temperatures recorded at the second and third heating run are shown in Fig. 6.

DSC curves of compositions E6/10 %DS1/Z-1 cured isothermally as recorded during second (a) and third (b) heating run
As expected, for the isothermally cured systems: E6/Z-1, E6/DS2, E6/10 %DS1/Z-1, and E6/10 %DS2/Z-1, all heated at the rate of 10 °C min−1 in the range of 0 through 250 °C, the lowest measured glass transition temperatures in the second heating run were those for the samples cured at the lowest temperature. The samples having the lowest T g were clearly not fully cured, as one could also guess examining the curing enthalpies (Table 4). These samples yielded also the highest post-curing exothermic effect starting already at the glass transition (Fig. 6a). Changes in the heat flow typical for pure glass transition were observed in the curves recorded in the third run.
For E6/Z-1 reference system cured isothermally, the glass transition temperatures recorded in the second run were only by 8–17 degrees higher than the curing temperature. The lower the curing temperature, the higher the differences between curing temperature and T g’s. Only after the third run, it became close to the highest value of 114 °C, recorded for the sample initially cured at 100 °C. All the epoxy polymers containing siloxane modifier had generally smaller values of T g, well seen for the samples cured isothermally at higher temperature and after the third heating run. At the presence of 10 % of siloxane modifiers, the highest attainable T g’s was by 14–17 °C lower than those for the reference system. This is an evident effect of the flexibility of siloxane molecules, since conversion of the system E6/10 %DS1/Z-1 was not lower than that for the reference one.
In the case of composition with 1,3-bis(aminopropyl)-1,1,3,3-tetramethyldisiloxane, a significant role seemed to play the change of cross-link density resulted from replacement of a part of hexafunctional curing agent Z-1 with tetrafunctional monomer DS2.
The lack of significant differences in T g of the systems with 10 % of DS1 or DS2 and that of the reference system cured isothermally at low temperatures was probably a result of higher conversion of siloxane modified system as compared to E6/Z-1, which compensated the flexibility effect of modifier. This effect obviously disappeared during the third heating run applied to siloxane modified systems.
The second heating run in the range of 0–250 °C applied to the compositions initially cured at low temperature did not bring about the value of glass transition temperature as high as for those cured at higher temperature. The latter reached higher conversion in the initial curing. Apparently, the fixed cross-linked structure of the systems cured at low temperature did not allow the unreacted groups to react during subsequent heating runs.
For the system E6/DS2 cured at 40 °C, the T g measured in the second heating run was 58 °C, whereas for the same system cured at 60 °C, it jumped to 72 °C. Further increase in isothermal curing temperature did apparently not improve the conversion; hence, the glass transition temperatures during the second and third heating run were similar (ranged from 76 to 81 °C) proving that curing temperature of 70 °C was sufficiently high for that system to reach full conversion.
No exothermic effect suggesting post-curing was observed for the system 1,3-bis(glycidyloxypropyl)-1,1,3,3-tetramethyldisiloxane/triethylenetetramine (DS1/Z-1) cured isothermally at temperatures above 60 °C. Clearly, the system was sufficiently flexible to reach full accessible conversion. All samples had elastomeric nature, and their T g ranged from −33 to −30 °C.
Conclusions
The reactivity of five epoxy compositions: E6/Z-1, DS1/Z-1, E6/DS2, E6/10 %DS1/Z-1, and E6/10 %DS2/Z-1 was determined both in isothermal conditions and at curing temperature changing linearly with time (dynamic conditions). The cured compositions were heated again to determine their glass transition temperature. The rate of temperature change during cross-linking was found not to affect the enthalpy of curing. However, the compositions cross-linked at small heating rate had higher glass transition temperature that those cured at a high heating rate. On the other hand, for the samples cured at isothermal conditions, the higher the curing temperature, the higher the enthalpy of cross-linking. The lowest activation energy was determined for the two-component systems where one of the monomer was a siloxane, i.e., DS1/Z-1 and E6/DS2. For the mixed systems containing 10 % of siloxane monomers, the activation energy was slightly smaller and the reaction time longer than those for the reference system E6/Z-1.
The best phenomenological model describing the curing of systems involving bisphenol A liquid epoxy resin E6 (see Table 1) was that of n-order autocatalytic reaction \( \left( {1 - \alpha } \right)^{\text{n}} \left( {1 + K_{\text{cat}} \alpha } \right) \) with n = 1.8–1.9 and \( \log K_{\text{cat}} \) = 0.6–0.8. The curing reaction of the system with siloxane epoxy monomer DS2/Z-1, on the other hand, was well described by the Prout–Tompkin equation: \( \left( {1 - \alpha } \right)^{\text{n}} \alpha^{\text{a}} \) with n = 1.3 and a = 0.37.
References
Pascault JP, Williams RJJ. Epoxy polymers. Weinheim: Wiley; 2010.
Jirasutsakul I, Paosawatyanyong B, Bhanthumnavin W. Aromatic phosphorodiamidate curing agent for epoxy resin coating with flame-retarding properties. Prog Org Coat. 2013;76:1738–46.
Zhang W, Li X, Jiang Y, Yang R. Investigations of epoxy resin flame-retarded by phenyl silsesquioxanes of cage and ladder structures. Polym Degrad Stab. 2013;98:246–54.
Chang HC, Lin HT, Lin CH, Su WC. Facile preparation of a phosphinated bisphenol and its low water-absorption epoxy resin for halogen-free copper clad laminates. Polym Degrad Stab. 2013;98:102–8.
Zhang W, Li X, Yang R. Blowing-out effect in epoxy composites flame retarded by DOPO–POSS and its correlation with amide curing agents. Polym Degrad Stab. 2012;97:1314–24.
Zhang W, Li X, Fan H, Yang R. Study on mechanism of phosphorus–silicon synergistic flame retardancy on epoxy resin. Polym Degrad Stab. 2012;97:2241–8.
Wang X, Song L, Pornwannchai W, Hu Y, Kandola B. The effect of graphene presence in flame retarded epoxy resin matrix on the mechanical and flammability properties of glass fiber-reinforced composites. Compos Part A. 2013;53:88–96.
Haurie L, Lacasta AM, Ciudad A, Realinho V, Velasco JI. Addition of flame retardants in epoxy mortars: thermal and mechanical characterization. Constr Build Mater. 2013;42:266–70.
Rosu D, Mustata F, Cascaval CN. Investigation of the curing reactions of some multifunctional epoxy resins using differential scanning calorimetry. Thermochim Acta. 2001;370:105–10.
Rosu D, Cascaval CN, Mustata F, Ciobanu C. Cure kinetics of epoxy resins studied by non-isothermal DSC data. Thermochim Acta. 2002;383:119–27.
Matějka L, Murias P. Control of the interphase interaction and morphology in the organic–inorganic polymer nanocomposites. J Mater Sci Eng. 2010;10:45–50.
Matějka L, Murias P, Pleštil J. Effect of POSS on thermomechanical properties of epoxy–POSS nanocomposites. Eur Polym J. 2012;48:260–74.
Matějka L, Strachota A, Pleštil J, Whelan P, Steinhart M, Šlouf M. Epoxy networks reinforced with polyhedral oligomeric silsesquioxanes (POSS). Structure and morphology. Macromolecules. 2004;37:9449–56.
Strachota A, Kroutilová I, Kovářová J, Matějka L. Epoxy networks reinforced with polyhedral oligomeric silsesquioxanes (POSS). Thermomechanical properties. Macromolecules. 2004;37:9457–64.
Boey FYC, Yap BH. Microwave curing of an epoxy–amine system: effect of curing agent on the glass-transition temperature. Polym Test. 2001;20:837–45.
Ramírez C, Rico M, Torres A, Barral L, López J, Montero B. Epoxy/POSS organic–inorganic hybrids: ATR–FTIR and DSC studies. Eur Polym J. 2008;44:3035–45.
Dai Z, Li Y, Yang S, Zhao N, Zhang X, Xu J. Kinetics and thermal properties of epoxy resins based on bisphenol fluorene structure. Eur Polym J. 2009;45:1941–8.
Okabe T, Takehara T, Inose K, Hirano N, Nishikawa M, Uehara T. Curing reactions of epoxy resin composed of mixed base resin and curing agent: experiments and molecular simulation. Polymer. 2013;54:4660–8.
Ooi SK, Cook WD, Simon GP, Such CH. DSC studies of the curing mechanisms and kinetics of DGEBA using imidazole curing agents. Polymer. 2000;41:3639–49.
Weibing X, Pingsheng H, Dazhu C. Cure behavior of epoxy resin/montmorillonite/imidazole nanocomposite by dynamic torsional vibration method. Eur Polym J. 2003;39:617–25.
Lin ST, Huang SK. Thermal degradation study of siloxane-DGEBA epoxy copolymers. Eur Polym J. 1997;33:365–73.
Hsiue GH, Wei HF, Shiao SJ, Kuo WJ, Sha YA. Chemical modification of dicyclopentadiene-based epoxy resins to improve compatibility and thermal properties. Polym Degrad Stab. 2001;73:309–18.
Lee SS, Kim SC. Morphology and properties of polydimethylsiloxane-modified epoxy resins. J Appl Polym Sci. 1997;64:941–55.
Ahmad S, Gupta AP, Sharmin E, Alam M, Pandey SK. Synthesis, characterization and development of high performance siloxane-modified epoxy paints. Prog Org Coat. 2005;54:248–55.
Murias P, Maciejewski H, Galina H. Epoxy resins modified with reactive low molecular weight siloxanes. Eur Polym J. 2012;48:769–73.
Kwon Y, Yim BS, Kim JM, Kim J. Mechanical and wetting properties of epoxy resins: amine-containing epoxy-terminated siloxane oligomer with or without reductant. Microelectron Reliab. 2011;51:819–25.
Yilgör E, Yilgör I. 1,3-bis(γ-aminopropyl)tetramethyldisiloxane modified epoxy resins: curing and characterization. Polymer. 1998;39:1691–5.
Park SJ, Jin FL, Park JH, Kim KS. Synthesis of a novel siloxane-containing diamine for increasing flexibility of epoxy resins. Mater Sci Eng. 2005;399:377–81.
Díaz I, Chico B, de la Fuente D, Simancas J, Vega JM, Morcillo M. Corrosion resistance of new epoxy–siloxane hybrid coatings. A laboratory study. Prog Org Coat. 2010;69:278–86.
Wang PI, Bult J, Ghoshal R, Ghoshal R, Lu TM. Rapid ultraviolet-curing of epoxy siloxane films. Mater Chem Phys. 2011;129:678–82.
Liu W, Qiu Q, Wang J, Huo Z, Sun H. Curing kinetics and properties of epoxy resin–fluorenyl diamine systems. Polymer. 2008;49:4399–405.
Yoo MJ, Kim SH, Park SD, Lee WS, Sun JW, Choi JH, Nahm S. Investigation of curing kinetics of various cycloaliphatic epoxy resins using dynamic thermal analysis. Eur Polym J. 2010;46:1158–62.
Saad GR, Elhamid EEA, Elmenyawy SA. Dynamic cure kinetics and thermal degradation of brominated epoxy resin–organoclay based nanocomposites. Thermochim Acta. 2011;524:186–93.
Wang H, Zhang Y, Zhu L, Du L, Zhang B, Zhang Y. Curing behaviors and kinetics of epoxy resins with a series of biphenyl curing agents having different methylene units. Thermochim Acta. 2011;521:18–25.
Maity T, Samanta BC, Dalai S, Banthia AK. Curing study of epoxy resin by new aromatic amine functional curing agents along with mechanical and thermal evaluation. Mater Sci Eng. 2007;464:38–46.
Ghaffari M, Ehsani M, Khonakdar HA, Assche GV, Terryn H. Evaluation of curing kinetic parameters of an epoxy/polyaminoamide/nano-glass flake system by non-isothermal differential scanning calorimetry. Thermochim Acta. 2012;533:10–5.
Cai H, Li P, Sui G, Yu Y, Li G, Yang X, Ryu S. Curing kinetics study of epoxy resin/flexible amine toughness systems by dynamic and isothermal DSC. Thermochim Acta. 2008;473:101–5.
Kim WG, Lee JY. Contributions of the network structure to the cure kinetics of epoxy resin systems according to the change of hardeners. Polymer. 2002;43:5713–22.
Hseih HK, Su CC, Woo EM. Cure kinetics and inter-domain etherification in an amine-cured phenoxy/epoxy system. Polymer. 1998;39:2175–83.
Corsetti RR, Neumeyer T, May M, Jandrey D, Altstaedt V, Cardozo NSM. Modeling and estimation of parameters for the curing of an epoxy/amine system. Polym Test. 2013;32:647–54.
Ramos JA, Pagani N, Riccardi CC, Borrajo J, Goyanes SN, Mondragon I. Cure kinetics and shrinkage model for epoxy–amine systems. Polymer. 2005;46:3323–8.
Li C, Fan H, Hu J, Li B. Novel silicone aliphatic amine curing agent for epoxy resin: 1,3-Bis(2-aminoethylaminomethyl) tetramethyldisiloxane. 2. Isothermal cure, and dynamic mechanical property. Thermochim Acta. 2012;549:132–9.
Gonzales M, Kadlec P, Štépánek P, Strachota A, Matéjka L. Crosslinking of epoxy–polysiloxane system by reactive blending. Polymer. 2004;45:5533–41.
Strachota A, Whelan P, Křiž J, Brus J, Urbanová M, Šlouf M, Matějka L. Formation of nanostructured epoxy networks containing polyhedral oligomeric silsesquioxanes (POSS) blocks. Polymer. 2007;48:3041–58.
Williams RJJ. Transitions during network formation. In: Pascault JP, Williams RJJ, editors. Epoxy polymers. Wiley: Weinheim; 2010.
Gillham JK. Cure and properties of thermosetting systems. Br Polym J. 1985;17:224–6.
Enns JB, Gillham JK. Time-temperature-transformation (TTT) cure diagram: modeling the cure behavior of thermosets. J Appl Polym Sci. 1983;28:2567–91.
Vyazovkin S, Burnham AK, Criado JM, Pérez-Maqueda LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Málek J. Kinetic analysis of crystallization processes in amorphous materials. Thermochim Acta. 2000;355:239–53.
Pilawka R, Mąka H. Kinetics of thermal decomposition of isocyanate–epoxy materials crosslinked in the presence of 1-ethylimidazole accelerator. Polimery (Warsaw). 2014;59:409–15.
Ozawa T. Kinetic analysis of derivative curves in thermal analysis. J Thermal Anal. 1970;2:301–24.
Kissinger HE. Reaction kinetics in differential analysis. Anal Chem. 1957;29:1702–6.
Koga N. Ozawa’s kinetics method for analyzing thermoanalytical curves. History and fundamentals. J Therm Anal Calorim. 2013;113:1527–41.
Šimon P. Isoconversional methods. Fundamentals, meaning and application. J Therm Anal Calorim. 2004;76:123–32.
Harsch M, Karger-Kocsis J, Holst M. Influence of fillers and additives on the cure kinetics of an epoxy/anhydride resin. Eur Polym J. 2007;43:1168–78.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J Polym Sci. 1964;C6:183–95.
Akahira T, Sunose T. Method of determining activation deterioration constant of electrical insulating materials. Res Rep Chiba Inst Technol (Sci Technol). 1971;16:22–31.
Cao J, Hua J, Fan H, Wan J, Li B. Novel silicone–phenyl contained amine curing agent for epoxy resin: 1. Non-isothermal cure and thermal decomposition. Thermochim Acta. 2014;593:30–6.
Acknowledgements
Financial support of Structural Funds in the Operational Program: Innovative Economy (IE OP) financed from the European Regional Development Fund, Project No. POIG.0101.02-00-015/08, is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Murias, P., Byczyński, Ł., Maciejewski, H. et al. A quantitative approach to dynamic and isothermal curing of an epoxy resin modified with oligomeric siloxanes. J Therm Anal Calorim 122, 215–226 (2015). https://doi.org/10.1007/s10973-015-4703-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4703-0