
Abstract
Thermal decomposition of suitable coordination compounds may be used as efficient route for fabrication of semiconducting layers. A new potential CdS precursor—a cadmium complex with all-sulfur Cd-coordination sphere [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1—has been prepared, and its properties are investigated. The complex was obtained in the reaction between dimeric bis(tri-tert-butoxysilanethiolato)cadmium(II) [Cd{SSi(OBut)3}2]2 and ammonium N,N-tetrametylene-dithiocarbamate and characterized by spectral methods (IR, UV–Vis, MS, and NMR). X-ray structure analysis revealed the complex as molecular and dimeric in solid state with each of chelating dithiocarbamate ligands bonded to one Cd center and sulfur atoms from two tri-tert-butoxysilanethiolato ligands bridging metallic centers and thus completing the CdS4 coordination sphere. Thin film of the precursor prepared on SiO2 substrates via spin-coating technique was analyzed by AFM. Its decomposition was studied by thermal analysis methods (TG, DSC, and TG-FTIR). After melting at 227 °C, [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 undergoes endothermic decomposition leading to CdS as the only solid product further identified by XRD, EDS, FIR as hexagonal CdS form. Its morphology is characteristic and may be described as “micro-noodles”.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The chemistry of complexes with S-donor ligands has recently gained renewed attention because of their use as efficient precursors of metal sulfides [1]. A special attention is paid for semiconductor nanocrystals due to their unique electronic and optical properties, and thus for their technological potential. CdS belongs to the II–VI compound family and is a well-studied semiconductor with a direct band gap of 2.42 eV and widespread applications, e.g., in solar cells, optical detectors, and optoelectronic devices [2]. It has also been a subject of intensive research not only because of its useful band gap but also high absorption coefficient, conversion efficiency, stability, and low cost [3]. CdS thin films are a desirable and the most commonly used window materials for high-efficiency polycrystalline thin-film photovoltaic devices [4, 5].
CdS layers can be obtained using the methods such as vacuum evaporation, chemical vapor deposition, spray pyrolysis, cathodic pulverization, sputtering, sintering, or screen printing [6]. Among these methods, pyrolysis is a simple, convenient, and low-cost technique for a large-area production. Although the obtained films are polycrystalline, stable, and uniform, still many efforts are devoted to the preparation of systems with the desired properties. It can be achieved, e.g., by controlling the size, morphology, and crystallinity of the products.
During the last two decades, the studies of metallic complexes with S-donors showed a progressive development. Species such as thiolates, dithiocarbamates (dtc) [7–11], dithiophosphinates [12], xanthates [13–15], and other compounds [16] have been successfully used to obtain respective sulfides. So far, dithiocarbamates remain most widely used for this purpose with dtc being undoubtedly the one of the most explored S,S′-chelate systems. We found it remarkable that despite vast knowledge of complexes with S-donor ligands, still there is very little known about the preparation, properties, and usage of systems containing simultaneously two different S-donors. For compounds with −S2CNR2 ligands we found that only one paper—concerning tin complexes with dithiocarbamato- and thiolato-rests ligating the same metallic centre [17]—falls within this subject.
We have recently reported synthesis of a first molecular Cd(II) trialkoxysilanethiolate bearing simultaneously dtc ligand [18] together with some studies concerning its thermal decomposition [19, 20]. We regard molecular silanethiolates as an alternative to conventional, much more explored and usually polymeric thiolates. Synthesis of heteroleptic complexes with this type of ligands and sulfur-rich metal coordination sphere seems to be a promising route to the preparation of species of greater volatility and desirable decomposition pathway.
Herein, we report on the preparation and properties of a new compound—a neutral, binuclear complex [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1 together with the data concerning its thermal decomposition and characterization of thus obtained CdS.
Experimental
Materials
All solvents were dried according to common procedures, degassed, and distilled prior to use. Dimeric bis(tri-tert-butoxysilanethiolato)cadmium(II) complex was prepared as described elsewhere [21]. Ammonium N,N-tetrametylenedithiocarbamate, C4H8NCS2NH4 (Fluka), and other commercially available chemicals were used as received.
Synthesis of complexes
Ammonium N,N-tetrametylenedithiocarbamate (0.049 g) was dissolved in 7 cm3 of distilled water and mixed with a solution of cadmium silanethiolate (0.135 g) dissolved in 10 cm3 of hot toluene. The content of the reaction flask was shaken vigorously for ca. 4 h using Universal Shaker 327 (Premed). Organic layer was separated, washed three times with distilled water, and dried over MgSO4 (Chempur). The resulting solution was evaporated to dryness, and the solid residue was recrystallized from toluene giving crystals of 1. Monocrystals of compound 1 of quality sufficient for X-ray measurements were obtained by dissolution of the complex in toluene at room temperature, followed by slow crystallization.
Yield (1) 75 %, Anal. Found: C, 37.92; S, 17.72; H, 6.70; N, 2.71 %. Calc. for C34H70O6N2S6Si2Cd (M r = 1076,33) C, 37.94; S, 17.87; H, 6.55; N, 2.60 %. IR (KBr pellet, cm−1): ν(C–O) 1179; ν(Si–O) 1072, 1047, 1028, 1002; ν (Si–S) 691, 633, 530. FIR (PS, cm−1): 243 (Cd–S). UV–Vis (CH3OH solution, nm): 205, 252, 285. 1H NMR (CDCl3), δ (ppm): 1.43 (s, 27H,CH 3 (But)), 2.08–2.13 (qui, 4H, CH2CH 2CH2), 3.7–3.78 (t, 4H, N-CH 2). MS, m/e: 539 [Cd{SSi(OBut)3}(S2CNC4H8)], 265(100) [HSSi(OBu)3-Me], 209(31) [(265)-C4H8], 153(93) [(209)-C4H8], 95(75) [HSSi(OH)2], 57(98) [C4H9].
Physical measurements
Elemental analysis
Elemental analysis of carbon, hydrogen, nitrogen, and sulfur was carried out by CHNS Euro-EA model 3018 (EuroVector) microanalyzer.
Spectroscopic measurements
IR spectra were recorded on a FTIR Mattson Genesis II Gold, externally controlled by WinFirst software. Spectra were registered in solid state (KBr pellet) and in solution (CCl4, KBr cuvette, 0.248 mm) in the range of 4000–400 cm−1. FIR spectra were recorded using BRUKER spectrometer IFS66. Measurements were made in polystyrene film with 0.12 cm−1 resolution in the range of 700–4.0 cm−1. UV spectra were recorded using Unicam UV 300 spectrometer externally controlled by Vision 32 software. Quartz cuvettes and methanol as solvent were used.
1H NMR spectra were registered with Gemini 200 (200 MHz) spectrometer (Varian) in CDCl3, and mass spectra were recorded using Finnigan Mat 95 (EI 75 eV).
Thermal analysis
Thermogravimetry coupled with IR was carried out in argon flow (purge: 90 mL min−1 and protective: 65 mL min−1) using Netzsch Thermobalance TG 209 coupled with a Bruker IFS66 FTIR spectrometer. Approximately, 8–10 mg samples were taken in Al2O3 crucibles. The experiments were carried out in the temperature range of 20–500 °C, for the heating rates of 2, 5, and 10 K min−1. The volatiles evolving from the heated sample were transported to the spectrometer chamber via thermostated pipe in a stream of argon.
Differential Scanning Calorimetry measurements were performed with the DSC 7 (Perkin–Elmer). Samples of ca. 5 mg, encapsulated in aluminum containers, were melted and recrystallized at 10 °C min−1 rate in an inert gas atmosphere (N2).
Spin-coating and characterization of films
Thin films of the complex were prepared on silicon oxide substrates via spin-coating (3000 RPM, 120 s.) using clear solutions of the complexes and the concentration of 1 mg mL−1 in toluene. Tapping mode AFM (Dimension 3100 Digital Instruments with Nanoscope IVa) was used to characterize the samples in terms of morphology.
Deposition of the complex
Samples of 1 were thermally treated in a furnace using a heating rate of 5 K min−1. The obtained yellow solid residue was further investigated by XRD, EDS, and SEM techniques.
Electron microscopy
The SEM experiments were performed using a combined Philips-FEI XL 30 ESEM system, at an acceleration voltage of 30 kV. The digitized images were recorded at different magnifications.
EDS, XPS
The EDS measurements were also performed using Philips ESEM XL 30 equipped with an energy-dispersive X-ray diffractometer, EDX/EDAX Sapphire®. XPS measurements were performed using a Kratos AXIS ULTRA instrument with a monochromated Al Kα X-ray source (1486.6 eV) operated at 15 mA emission current and 10 kV anode potential. The instrument was used in FAT (fixed analyzer transmission) mode with a pass energy of 80 eV for wide scans and a pass energy 20 eV for high-resolution scans.
XRD
The X-ray patterns of the crystalline residues were recorded at room temperature using X’Pert Philips diffractometer (source radiation: Cu Kα1, λ = 0.1546 nm, 40 kV, 30 mA). For characterization and qualitative analysis, the results were compared with standard data from the International Centre for Diffraction Data [22].
Crystal structure determination
Suitable single crystal of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 was selected and mounted on a quartz capillary with epoxy resin. Experimental intensity data were collected at 295(2) K on a KM4 diffractometer, having goniometer equipped with Sapphire 2 CCD detector (Oxford Diffraction) with monochromated MoKα radiation (λ = 0.71073 Å). Data were collected in four series of 152 frames each, at φ = 0°, 90°, 180°, and 270°; ω scan width of 0.75°; and exposure time of 50 s per frame. The unit cell dimensions, additional crystallographic data, and refinement results for the complex are given in Table 1.
The structure was solved by direct methods and refined by least-squares calculation using anisotropic approximation for non-hydrogen atoms. H atoms were added at ideal positions and refined using the standard riding model. The thiocarbamate (S2CNC4H8) group was refined with C2–C5 atoms disordered over two positions occupied with probabilities 0.66(3)/0.34(3).
Data collection and reduction, as well as refinement of unit cell parameters, were performed using CrysAlis CCD and CrysAlis RED [23]. The structure solution and refinement (by full-matrix least-squares against F 2) was performed using WinGX program package (ver.1.80.05, Farrugia 1999) [24].
Results and discussion
In order to obtain complexes that can serve as precursors of respective sulfides, we have focused our attention on the synthesis of compounds with different S-donors (among them one with S–Si bond) ligating to the same metallic center. Our recent investigations [18] have proven that [Cd{SSi(OBut)3}2]2 [21]/(−)S2CNR2(dtc)/R′4NX reaction system can be used for this purpose. We noted, however, that besides the desired neutral complexes with sulfur-rich kernel, it was also possible to obtain ionic by-products, e.g. (Bu4N)2[CdBr4]·2C7H8 [25] and [Cd{SSi(OBut)3}(S2CNC4H8)I][Bu4N] [26] by just a small modification of the reaction system like a change of the solvent or quaternary salt used. Nevertheless, the reaction between [Cd{SSi(OBut)3}2]2 and C4H8NCS2NH4, carried out in the toluene/water mixture in the presence (or without) of tetra-n-butylammonium bromide, yielded plate-like colorless crystals of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1.
Molecule of 1 is electrically neutral centrosymmetric dimeric complex crystallizing in monoclinic system in C2/c space group. Each of its cadmium atoms is coordinated by four S atoms originating from one chelating dithiocarbamato- and two silanethiolato-rests. Both silanethiolate sulfur atoms act as bridges between two Cd atoms forming the planar central [2Cd–2S] ring. Because of the chelating character of dithiocarbamato ligands, the tetrahedral geometry at Cd atoms is severely distorted. Noticeable and favorable feature of 1 is the lack of additional intermolecular interactions within the crystal, as these can influence the melting point and volatility of the compound (Fig. 1).

Crystal and molecular structure of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1. Thermal ellipsoids 30 %. Carbon atoms C2–C5 are disordered over two positions occupied with probabilities 0.66(3)/0.34(3). For clarity, only 66 % disorder component is shown, and H atoms have been omitted. [Symmetry code: (a) −x, −y, −z]. Selected bond lengths (Å): Cd1–S1 2.614(1), Cd1–S1a 2.545(1), Cd1–S2 2.536(2), Cd1–S3 2.599(2), S2–C1 1.718(5), S3–C1 1.707(5), Si1–S1 2.1244(14) and angles (°): Cd1–S1–Cd1a 83.97(3), S1–Cd1–S1a 96.03(3), S1–Cd1–S2 131.46(5), S1a–Cd1–S3 130.51(5), S2–Cd1–S3 70.53(4), S2–C1–S3 120.0(3), Cd1–S1–Si1 93.88(5), Cd1–S2–C1 85.48(2), Cd1–S3–C1 83.71(2)
The thin film of the complex 1 was prepared via spin-coating on hydrophilic surface, i.e., native oxide-terminated Si(111), to examine nascent morphology of the thus obtained layer. The particle size and self-organization have been analyzed by AFM and TEM techniques. The AFM measurements performed in the tapping mode (Fig. 2) revealed that 1, when spin-casted from THF, assembled into porous layers with the pore diameters ranging from 50 to 500 nm. The thickness of the film is 3 nm. TEM images revealed also the existence of small (ca. 3 nm) particles.

AFM surface topography images of thin layer of complex 1 spin-cast on Si
The chemical composition of the spin-coated layers was determined quantitatively by means of X-ray photoelectron spectroscopy (XPS). Figure 3 displays the XPS spectrum of 1 clearly showing the presence of O, S, Cd and Si signals.

XPS spectrum of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1
Thermal analysis and complementary techniques
To assess the potential of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 as a convenient source of cadmium sulfide, its thermal decomposition has been investigated by a combination of DSC, TG, and IR techniques. Figure 4 shows the TG and DTG curves of the investigated compound recorded at 10 K min−1; similar curves were obtained for other heating rates (2, 5, 15 and 20 K min−1). Between 100 and 225 °C, the system loses around 4 % of its total mass, what most probably corresponds to that of the adsorbed solvent. Thermal decomposition of 1 starts at 229 °C that is somewhat characteristic for dimeric silanethiolate complexes of cadmium [19]. As shown in Fig. 4, thermal degradation of heteroleptic complex 1 proceeds in two steps (with DTGmax at 318.9 and 388 °C). The decomposition of homoleptic cadmium(II) tri-tert-butoxysilanethiolate and heterooleptic [Cd{μ-SSi(OBut)3}(S2CNEt2)]2 [18] starts within the same temperature but proceeds in only one step. Therefore to obtain decomposition product of 1 the annealing should be carried up to at least 400 °C, which is nearly 90 °C higher than that for [Cd{SSi(OBut)3}2]2 and 50 °C higher than that for [Cd{μ-SSi(OBut)3}(S2CNEt2)]2.

Thermal decomposition curves of the homoleptic substrate [Cd{SSi(OBut)3}2]2 and heteroleptic [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1 (heating rate 10 K min−1)
After melting at 227 °C, [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 undergoes endothermic decomposition between 260 and 325 °C. The DSC curve indicates two heat transfers, corresponding to the melting and decomposition of the investigated compound (Fig. 5).

Differential scanning calorimetry curves recorded for [Cd{SSi(OBut)3}(S2CNC4H8)]2 (at the heating rate of 10 K min−1)
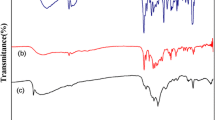
Figure 6 shows TG-FTIR spectra recorded for products evolved during thermal decomposition of complex 1. Two major groups of species may be identified related to S2CNC4H8 and tri-tert-butoxysilyl substituents, respectively, with the latter, most probably undergoing further decomposition into gaseous products possessing both Si–OH (v(Si–O) 1075 cm−1) and Si-OBut (v(C–O) 1190 cm−1) fragments. The presence of bands arising from symmetric and asymmetric scissor-bending vibration modes characteristic for tert-butyl substituent (1392 and 1369 cm−1) as well as skeleton vibration of -But rest as a whole (1242 cm−1) is noteworthy. In the FT-IR spectra of the off-gases, a small amount of ethyl isothiocyanate (C2H5NCS) with absorption bands at 2073 and 2051 cm−1 can also be seen (Fig. 7).

3D IR spectra of volatiles evolving during TG analysis in the function of temperature for [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2

Selected IR spectra of volatiles evolving during TG analysis of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 recorded at several temperatures for the heating rate of 10 K min−1
The introduction of dithio- substituents can thus modify the decomposition character, and the decomposition scheme of 1 can be tentatively described in Scheme 1.

Decomposition scheme
This is in good agreement with data obtained from the TG experiment (Tables 2, 3)
The characterization of the decomposition product
The final product of decomposition process (obtained when the sample was heated to 410 °C) is CdS, as confirmed by FIR (ν Cd–S at 254 cm−1, Fig. 8) and XRD analysis.

Far infrared spectrum of the final product of decomposition of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 1
To assess the potential of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 as a source of cadmium sulfide, we have also decomposed this compound using the pyrolysis deposition technique and obtained thin films of yellow product deposited on a glass substrate. The X-ray pattern of the residue indicated only the presence of α-CdS wurzite-type crystallites (Fig. 9). For characterization and qualitative analysis, the results were compared with standard data from the International Centre for Diffraction Data [22]. Additional EDS measurement revealed that the decomposition leads to CdS as the only deposited product (sulfur K line at 2.3076 keV; cadmium L line at 3.1315 keV as seen in Fig. 10).

XRD pattern for the decomposition product of [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2

EDS spectrum recorded for polycrystalline material obtained by pyrolysis of the complex [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2
Cadmium sulfide obtained by the thermal decomposition of 1 possesses an interesting morphology as shown in Fig. 11a–d. SEM images reveal that it consists of large quantity of structures which can be best described as “micro-noodles” of ca. 500 nm wide and several micrometers long. For CdS, no such data were yet reported. Similar shapes were observed, e.g., by O’Brien and coworkers in the case of NiS [27].

SEM images of CdS obtained using [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 as precursor
Conclusions
In conclusion, we confirmed our earlier observations that a reaction of dimeric bis(tri-tert-butoxysilanethiolato)cadmium(II) [Cd{SSi(OBut)3}2]2 with dithiocarbamate ammonium salts results in the formation of heteroleptic cadmium(II) complexes. The crystals of stable heteroleptic [Cd{μ-SSi(OBut)3}(S2CNC4H8)]2 complex are formed of binuclear molecules lacking secondary intermolecular interactions. The thermal decomposition of 1 proceeds in two steps. As indicated by the TG experiments, the complex possesses sufficient volatility, and above ca. 300 °C, undergoes thermal decomposition forming yellow crystalline deposits. The X-ray pattern of the residue indicates only the presence of α-CdS. The most notable feature is an interesting and reproducible “micro-noodleslike” morphology of the thus obtained deposits. From the application perspective (thermal treatment), the complex fulfills the primary requirements necessary for a cadmium sulfide precursor.
References
McCleverty JA, Meyer TJ, editors. Comprehensive coordination chemistry II. From biology to nanotechnology. Amsterdam: Elsevier; 2003.
Ruxandra V, Antohe S. The effect of the electron irradiation on the electrical properties of the polycrystalline CdS thin layers. J Appl Phys. 1998;84:727–33.
AlKuhaimi SA. Influence of preparation technique on the structural, optical and electrical properties of polycrystalline CdS films. Vacuum. 1998;51:349–55.
Valyomana AG, Vijayakumar KP, Purushothaman C. Conductivity studies on spray-pyrolysed CdS films in ambient conditions. J Mater Sci Lett. 1992;11:616–8.
Őzsan ME, Johnson DR, Sadeghi M, Svapathasundaram D, Godlet G, Furlong MJ, Peter LM, Shingleton AA. Optical and electrical characterization of CdS thin films. J Mater Sci Mater Electron. 1996;7:119–25.
Esteves ACC, Trindade T. Synthetic studies on II/VI semiconductor quantum dots. Curr Opin Solid State Mater Sci. 2002;6:347–53.
Fainer NI, Rumyantsev YuM, Salman EG, Kosinova ML, Yurjew GS, Sysoeva NP, Maximovskii EA, Sysoev SA, Golubenko AN. Plasma-deposited CdS layers from (o-phen) bis (diethyldithiocarbamate) cadmium. Thin Solid Films. 1998;286:122–6.
Fainer NI, Kosinova ML, Rumyantsev YM, Saman EG, Kuznetsov FA. Growth of PbS and CdS thin films by low-pressure chemical vapour deposition using dithiocarbamates. Thin Solid Films. 1996;280:16–9.
O’Brien P, Walsh JR, Watson IM, Hart L, Silva SRP. Properties of cadmium sulphide films grown by single-source metalorganic chemical vapour deposition with dithiocarbamate precursors. J Cryst Growth. 1996;167:133–42.
Coi I-H, Yu PY. Structural and optical properties of cubic-CdS and hexagonal-CdS thin films grown by MOCVD on GaAs substrates using a single-source precursor C14H30CdN2S4. Phys Status Solidi B. 2005;242:1610–6.
Zhang YC, Chen WW, Hu XY. Controllable synthesis and optical properties of Zn-doped CdS nanorods from single-source molecular precursors. Cryst Growth Des. 2007;7:580–6.
Byrom C, Malik MA, O’Brien P, White AJP, Williams DJ. Synthesis and X-ray single crystal structures of bis(diisobutyldithiophosphinato)cadmium(II) or zinc(II): potential single-source precursors for II/VI materials. Polyhedron. 2000;19:211–5.
Nair PS, Radhakrishnan T, Revaprasadu N, Kawole G, O’Brien P. Cadmium ethylxanthate: a novel single-source precursor for the preparation of CdS nanoparticles. J Mater Chem. 2002;12:2722–5.
Barreca D, Gasparotto A, Maragno C, Seraglia R, Tondello E, Venzo A, Krishnan V, Bertagnolli H. Cadmium O-alkylxanthates as CVD precursors of CdS: a chemical characterization. Appl Organomet Chem. 2005;19:59–67.
Cusack J, Drew MGB, Spalding TR. Syntheses and spectroscopy of diamine complexes of Zn(II) and Cd(II) ethylxanthates and the molecular structures of [M(S2COEt)2TMEDA]: formation of CdS nanoparticles from [Cd(S2COEt)2] and [Cd(S2COEt)2TMEDA]. Polyhedron. 2004;23:2315–21.
Nair PS, Radhakrishnan T, Revaprasadu N, Kolawole GA, O’Brien P. Cd(NH2CSNHNHCSNH2)Cl2: a new single-source precursor for the preparation of CdS nanoparticles. Polyhedron. 2003;22:3129–35.
Barone G, Chaplin T, Hibbert TG, Kana AT, Mahon MF, Molloy KC, Worsley ID, Parkin IP, Price LS. Synthesis and thermal decomposition studies of homo- and heteroleptic tin (IV) thiolates and dithiocarbamates: molecular precursors for tin sulfides. Dalton Trans. 2002;6:1085–92.
Kropidłowska A, Chojnacki J, Fahmi A, Becker B. In search of molecular precursors for cadmium sulfide—new complexes with sulfur-rich kernel: cadmium(II)tri-tert-butoxysilanethiolates with additional diethyldithiocarbamato ligand. Dalton Trans. 2008;47:6825–31.
Kropidłowska A, Rotaru A, Strankowski M, Becker B, Segal E. Thermal stability and non-isothermal decomposition kinetics of a heteroleptic cadmium(II) complex, potential precursor for semiconducting CdS layers. J Therm Anal Calorim. 2008;91:903–9.
Rotaru A, Mietlarek-Kropidłowska A, Constantinescu C, Scariooreanu N, Dumitru M, Strankowski M, Rotaru P, Ion V, Vasiliu C, Becker B, Dinescu M. CdS thin films obtained by thermal treatment of cadmium(II) complex precursor deposited by MAPLE technique. Appl Surf Sci. 2009;255:6786–9.
Wojnowski W, Becker B, Walz L, Peters K, Peters EM, von Schnering HG. Contributions to the chemistry of silicon-sulphur compounds—LX. Synthesis, molecular structure and properties of the dimeric cadmium(II) bis(tri-tert-butoxysilanethiolate)(t-C4H9O)3SiS2Cd2. Polyhedron. 1992;11:607–12.
ICDD PDF-2. Database Release 1998. ISSN 1084-3116.
Oxford Diffraction. CrysAlis CCD and CrysAlis RED, version 1.171.27p5. Abingdon: Oxford Diffraction Ltd; 2004.
Farrugia LJ. WinGX suite for small-molecule single-crystal crystallography. J Appl Cryst. 1999;32:837–8.
Kropidłowska A, Chojnacki J, Becker B. Bis(tetra-n-butyl-ammonium)tetra-bromocadmate(II) toluene disolvate. Acta Cryst E. 2006;62:m457–9.
Kropidłowska A, Chojnacki J, Becker B. Tetra-n-butylammonium iodido-(pyrrolidine-1-carbodithioato-κ2S, S’)(tris-tert-butoxysilanethiolato-κS)cadmate(II). Acta Cryst E. 2008;64:m832.
O’Brien P, Park JH, Waters J. A single source approach to deposition of nickel sulfide thin films by LP-MOCVD. Thin Solid Films. 2003;431–432:502–5.
Acknowledgements
The authors gratefully acknowledge the financial support from the Polish Ministry of Science and Higher Education—Project Nr N N204 543339 and N N204 150237
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Mietlarek-Kropidłowska, A., Chojnacki, J., Strankowski, M. et al. Cadmium complex possessing simultaneously silanethiolato- and dithiocarbamato-ligands. A novel single-source precursor of cadmium sulfide. J Therm Anal Calorim 118, 993–1001 (2014). https://doi.org/10.1007/s10973-014-3842-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-014-3842-z