
Abstract
The objective of this work is to analyse the chemical stability of BaCe0.85Y0.15O3−δ –Ce0.85Y0.15O2−δ (BCY15–YDC15) composite materials at 600 °C and to compare the aforementioned chemical stability with that of pure BCY15. The composite powders were obtained by mixing together powders of BCY15 and YDC15 in the following volume fractions: 90 % BCY15 + 10 % YDC15, 70 % BCY15 + 30 % YDC15, 30 % BCY15 + 70 % YDC15, 20 % BCY15 + 80 % YDC15 and 10 % BCY15 + 90 % YDC15. After that both powders and sintered samples of the BCY15 and the BCY15–YDC15 composites were saturated in two different atmospheres at 600 °C: CO2/H2O (3.1 mol% H2O) and N2/H2O (46.8 mol% H2O). The effects of the previously mentioned atmospheres on the physicochemical properties of the samples were investigated via differential thermal analysis (DTA) combined with thermogravimetric analysis (TG). Furthermore, mass spectrometry was used to analyse the chemical composition of the gases released from the samples during the DTA–TG heating process. The surface and cross-section morphology of the samples were examined by scanning electron microscopy. Moreover, the phase composition of each sample was studied via X-ray Diffraction. From the combined analysis, it can be concluded that the addition of YDC15 in the composite samples leads to an increase in resistance against the corrosive effects of CO2. Furthermore, it was determined that all samples maintain stability in the presence of H2O at 600 °C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Barium cerate (BaCeO3) doped with trivalent cations was originally studied as a proton conducting electrolyte material by Iwahara et al. [1]. Since then, it has been determined that BaCe1−x Y x O3−δ (BCY) materials possess relatively high conductivity among competitors at intermediate temperatures (600–800 °C) [2–5]. Furthermore, it was determined that 15 % of the Y3+ dopant in BCY is the optimal amount for maximum conductivity at 600 °C [5]. Unfortunately, BaCeO3 has the tendency to react with CO2 forming barium carbonate (BaCO3) and cerium dioxide (CeO2), as shown in Eq. (1) [6]. BaCeO3 can also react with H2O, according to Eq. (2) [7]:
However, after exposing a BaCeO3-based material to an atmosphere containing 50 vol% H2O for 1,000 h at 600 and 700 °C, Wu and Liu [8] did not detect any amounts of Ba(OH)2 or CeO2 formed in the sample. This suggests that reaction (2) is not significant in the temperature range 600–700 °C for such materials. As for reaction (1), studies have shown that BaCe0.9Y0.1O2.95 can carbonate at temperatures below 700 °C even at low CO2 partial pressures [9].
There have been several attempts to improve the chemical stability of BaCeO3-based materials, such as substituting cerium with zirconium [10–14] or titanium cations [15, 16]. Studies have also shown that BaCeO3 doped with 30 % indium exhibits adequate chemical stability against CO2 compared with the traditional rare earth doped BaCeO3 [17]. In this work, it is assumed that a mixture of BaCe0.85Y0.15O3−δ (BCY15) and a CeO2-based material will exhibit greater chemical stability in the presence of both CO2 and H2O than pure BCY15. Ce0.85Y0.15O2−δ (YDC15) was chosen as the CeO2-based compound, because BCY15 and YDC15 have been determined as a priori compatible materials [18]. The theory behind the addition of YDC15 improving the chemical stability of BCY15 is that the presence of CeO2 doped with Y2O3 in the BCY15–YDC15 composites should shift the equilibrium in Eqs. (1) and (2) towards the substrates according to the Le Chatelier–Braun principle.
The above-mentioned composites are intended for use as potential central membrane (CM) materials in a new type of fuel cell operating in the temperature range 600–700 °C, entitled IDEAL-Cell for ′Innovative Dual mEmbrAne fueL Cell′ [19]. The basic idea behind the IDEAL-Cell is to completely eliminate the presence of H2O at one of the electrodes and therefore avoid an overall loss of fuel cell efficiency [19]. The concept behind an IDEAL-Cell, shown schematically in Fig. 1 [18, 19], lies in the junction between the anode/electrolyte (anode compartment) of a Protonic Ceramic Fuel Cell (PCFC) and the cathode/electrolyte (cathode compartment) of a Solid Oxide Fuel Cell (SOFC) via a mixed proton (H+) and oxide ion (O2−) conducting porous CM, from which water vapour is evacuated through open porosity [18–20].

The goal of this work is to determine the resistance of BCY15–YDC15 composite materials mixed together in different ratios against the corrosive effects of both CO2 and H2O at 600 °C and compare the chemical stability of the materials with that of pure BCY15 to confirm the effect of YDC15 addition on BCY15.
Experimental procedure
Materials and synthesis of samples
The initial BCY15 and YDC15 powders were synthesised via co-precipitation, in which barium nitrate Ba(NO3)2 (QUALITY CHEMICALS, 99.9 %), cerium nitrate Ce(NO3)2 (ALTICHEM, ≥99.5 %) and yttrium nitrate Y(NO3)3 (ALTICHEM, 99.99 %) were mixed together in the appropriate ratios with the precipitating agent ammonium carbonate (NH4)2CO3 (ALTICHEM, 99.5 %) to yield the desired precursors of BCY15 and YDC15, respectively. After drying and grinding the aforementioned precursors, the BCY15 precursor was given heat treatment at 1,000 °C for 6 h (heating/cooling rate: 100° h−1), whereas heat treatment for the YDC15 precursor was carried out at 900 °C for 1 h (heating/cooling rate: 300° h−1). In both cases, the obtained oxides underwent wet desagglomeration, drying and sieving, after which the compounds were thermally treated at 600 °C. As a result, the finished products (BCY15 and YDC15 powders) were achieved.
BCY15–YDC15 composite powders were obtained by mixing together the BCY15 and YDC15 powders in the ratios presented in Table 1 via ball milling (24 h, 120–150 rpm) in a polyethylene terephthalate (PET) bottle (vol 100 mL) in presence of zirconia (ZrO2) balls (4 balls with diameter = 15 mm, 34 balls with diameter = 10 mm and 98 g of balls with diameter = 5 mm) by adding isopropanol (≈20 mL). Then the mixtures were freeze-dried in liquid nitrogen and finally the mixtures were separated from ZrO2-balls by sieving (53 μm mesh).
In order to eliminate all impurities, BCY15 and BCY15–YDC15 composite powders were annealed at 1,100 °C for 2½ h in flowing synthetic air with a heating/cooling rate of 10 °C min−1. After annealing, the powders were pressed at 25 MPa into pellets of 5 mm diameter and 2 mm thickness. These pellets were then sintered at 1,200 °C for 2 h in flowing synthetic air. The rate of heating was 2.5 °C min−1 and cooling was 5 °C min−1.
Chemical stability and physicochemical property analysis
In order to expose both BCY15 and BCY15-YDC15 powders and sintered samples to CO2/H2O (3.1 mol% H2O) and N2/H2O (46.8 mol% H2O) atmospheres, the studied samples were entered into the system schematically, shown in Fig. 2, for 600 h. First, the studied samples were placed inside quartz crucibles and then entered into a reaction tube. The crucibles were lowered until they were located at the isothermal section of the furnace. Additionally, a thermocouple was used to ensure that the temperature in the isothermal section remains at 600 °C for the entire duration of the experiment (600 h). The desired gas mixture was obtained by supplying gas (CO2 or N2) to a saturator with a flow rate of 50 cm3 min−1, as determined by the mass flow controller. The amount of water vapour captured by the incoming gas was determined by the temperature of the water in the saturator. The desired water temperature, 25 °C in the case of CO2 and 80 °C in the case of N2, was achieved with the help of a heating element. The obtained gas mixture (CO2/H2O or N2/H2O) then entered into the reaction tube.

Scheme of the system used to saturate the samples in CO2/H2O or N2/H2O atmosphere at 600 °C for 600 h
After exposure to the above-mentioned conditions, the samples were heated to 1,200 °C with a rate of 20 °C min−1 in synthetic air flowing at 100 cm3 min−1. The effects of both CO2/H2O and N2/H2O at 600 °C on the physicochemical properties of the samples were determined during the heating process by a microthermogravimetric apparatus (SDT-2960 by TAInstruments, USA), which carried out combined Differential Thermal Analysis (DTA) and Thermogravimetric Analysis (TG). A quadruple mass spectrometer (Thermostar by Balzers Instruments, Liechtenstein) was also used for Mass Spectrometry (MS) studies of the chemical composition of the gases released from the samples during heating.
Microphotographs of the initial BCY15 and YDC15 samples, as well as the surface morphologies of the bulk samples after exposure to both N2/H2O and CO2/H2O, were obtained by Scanning Electron Microscopy (SEM) using a FEI NOVA NanoSEM 200 microscope. Additionally, a mercury intrusion porosimeter (PoreMaster 60 by Quantachrome Instruments, USA) was used to study the open porosity of the bulk samples. Initially, the samples were entered into a low pressure station to identify pores up to 10.66 μm in size. After that the analysis was carried out in a high pressure station capable of inserting mercury into pores between 10.66 μm and 3.60 nm.
Particle size distribution of the powders, expressed in volume fractions, was investigated via a Malvern Mastersizer 2000 particle size analyser with the sample preparer Hydro 2000S. The measurements were carried out for wet samples (in water solvent) with 100 % ultrasonic irradiation and a pump speed of 3,000 rpm. The measuring range of this equipment is between 0.02 and 2,000 μm, and the refractive index of the light used in the experiment is between 0.01 and 1.52. Moreover, the phase compositions of the composite samples and the initial powders were determined via X-ray Diffraction (XRD), which was carried out by an X‘Cellerator’ strip detector and an X`Pert Pro MPD diffractometer (PANalytical, USA) using monochromatic CuKα radiation.
Results and discussion
Initial powder characterisation
SEM microphotographs of the initially received BCY15 and YDC15 powders are shown in Fig. 3a, b, respectively. From these figures it follows that the BCY15 powder consists mostly of small non-agglomerated grains. On the other hand, larger aggregates are present in the YDC15 powder. This is confirmed by the cumulative particle size distribution analysis of the YDC15 powder (Fig. 4), from which it follows that 10 % vol of the grains is larger than D V90 = 2.27 μm, 10 vol% is smaller than D V10 = 0.15 μm and the volume median diameter of the grains is D V50 = 0.21 μm.

SEM microphotographs of the initial a BCY15 and b YDC15 powders

Particle size distribution analysis (in volume fractions) of the initial YDC15 powder
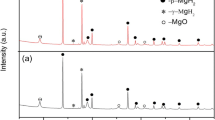
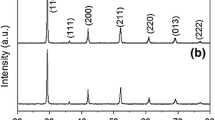
The XRD analysis of the initial BCY15 powder (Fig. 5a) detected two phases: BaCeO3 in which Ce4+ cations are partially substituted by Y3+ cations and small amounts of BaCO3 impurity. On the other hand, Fig. 5b shows that the initial YDC15 powder consists only of a single phase, CeO2 in which Ce4+ cations are also partially substituted by Y3+ cations. The elevation at positions from 10° to below ~38° in the diffraction patterns is the result of using a fixed divergence slit for these studies.

XRD patterns of a BCY15 and b YDC15 initial powders
Thermal analysis (DTA and TG) combined with MS studies
TG–MS studies of the powders after exposure to N2/H2O atmosphere at 600 °C for 600 h are shown in Fig. 6. From this figure, it follows that H2O has the largest effect on the BCY15 sample. Moreover, desorption of CO2 from the surface of the sample is determined during the heating process. As for the composite powders, up to 200 °C, very negligible amount of water vapour desorbed from the samples as confirmed by MS analysis. Beyond 700 °C, MS and TG curves indicate that mass loss occurs from decomposition of baseline amounts of BaCO3 present in the samples due to manipulation of the samples in air. Between 200 and 700 °C, the cause of the very minor mass changes in the samples has not been determined. It is possible that oxygen from the flowing synthetic air used during MS–TG analysis diffuses into the samples during heating. Unfortunately, signals from such minor oxygen changes in synthetic air atmosphere (~21 vol% O2) would be practically invisible in the MS analysis and therefore the theory remains unconfirmed. For each of the powders, total mass loss does not exceed 1 mass% and in the case of bulk samples saturated in N2/H2O atmosphere at 600 °C for 600 h, the total mass loss is less than 0.25 mass% for each material, as confirmed by TG studies presented in [21].

MS and TG curves of a BCY15, B90Y10 and B70Y30 and b Y90B10, Y80B20 and Y70B30 powders after ageing in N2/H2O rich atmosphere at 600 °C for 600 h
The results of the DTA–TG–MS analysis of the powders after saturation in CO2/H2O atmosphere at 600 °C for 600 h are presented in Fig. 7. From this figure, it follows that mainly CO2 was adsorbed during exposure to the above-mentioned conditions. Only minor effects corresponding to desorption of H2O were determined at temperatures up to 200 °C by MS and TG. The more visible DTA–TG effects, which take place above 700 °C, can be contributed to two-stage decomposition of BaCO3. This is further confirmed by the sudden increases in the amount of CO2 detected by MS in the same temperature range.

MS, DTA and TG curves of a BCY15, B90Y10 and B70Y30 and b Y90B10, Y80B20 and Y70B30 powders after ageing in CO2/H2O rich atmosphere at 600 °C for 600 h
In the case of the sintered samples exposed to the same conditions as mentioned above (Fig. 8), two stages of desorption are observed. The first takes place up around 200 °C and can be contributed to desorption of small amounts of H2O. The second stage, which corresponds to BaCO3 decomposition, is observed beyond 700 °C. In this case, one-stage BaCO3 decomposition is determined for all samples. Furthermore, the MS peak correlating to CO2 desorption from BCY15 is not as sharp as in the case of the BCY15 powder previously exposed to the same conditions. This can be explained as CO2 becoming trapped inside the grain boundaries and, as a result, the release of CO2 from the sample turning into a slow diffusion process. This phenomenon is not observed in the case of the composite bulk samples. In both figures (Figs. 7, 8), the mass loss is mostly caused by BaCO3 decomposition, whereas the effects of H2O on the samples are negligible.

MS, DTA and TG curves of: a BCY15, B90Y10 and B70Y30 and b Y90B10, Y80B20 and Y70B30 bulk samples after ageing in CO2/H2O rich atmosphere at 600 °C for 600 h
The approximate conversion of the powders and bulk samples into BaCO3 after saturation in CO2/H2O rich atmosphere at 600 °C for 600 h is compared in Fig. 9. From this figure, it follows that the correlation between conversion into BaCO3 and the amount of YDC15 in the powder is almost linear. This confirms the theory of YDC15 retarding the formation of BaCO3 in the samples. However, in the case of the bulk samples the correlation is different. The lowest percentage of barium was converted into BaCO3 in the BCY15 sample and the highest conversion was observed for B70Y30. This phenomenon can be explained by examining the porosities of the samples, the values of which are presented in Table 2. From these results, it follow that BCY15 has the lowest porosity among the studied materials. B90Y10 has a smaller amount of YDC15 than B70Y30 to prevent the formation of BaCO3. However, B70Y30 has higher porosity, which increases the rate of CO2 adsorption compared to that of B90Y10. The highest porosities among the materials were determined for the composite samples with more YDC15 than BCY15. The fact that a lower percentage of barium was converted into BaCO3 in those materials than in the case of B70Y30 can be attributed to the influence of larger amounts YDC15 in the samples.

Correlation between the approximate molar percentage of the conversion of barium into BaCO3 in the studied materials after exposure to CO2/H2O rich atmosphere at 600 °C for 600 h and the volume fractions of the initial BCY15 and YDC15 powders used to obtain the samples
XRD analyses of the phase composition of Y70B30 after saturation for 600 h at 600 °C in N2/H2O rich atmosphere and CO2/H2O rich atmosphere are shown in Fig. 10a, b, respectively. From Fig. 10a, it follows that after ball milling composites consisting of two phases are obtained from the initial one-phase powders. It can also be concluded that no additional phases were formed during exposure to N2/H2O atmosphere at 600 °C. On the other hand, the effects of CO2/H2O atmosphere at 600 °C are clearly seen in Fig. 10b, which confirms the formation of BaCO3 in the material. The baseline/signal ratio in Fig. 10b is very high, because the XRD studies of the composites after saturation in CO2/H2O were carried out on a small piece with an uneven surface after SEM analysis of the fracture cross-section. Graphite was also detected on the sample, because carbon was deposited on the surface of the sample to obtain a better SEM image.

XRD patterns of Y70B3O after saturation at 600 °C for 600 h in a N2/H2O rich atmosphere and b CO2/H2O rich atmosphere
The results of the SEM analysis show significant microfractures are not visible in the samples after prolonged exposure to N2/H2O or CO2/H2O rich atmosphere. SEM microphotographs of B70Y30 after saturation N2/H2O rich atmosphere and CO2/H2O atmosphere are presented as an example in Fig. 11a, b, respectively. The surface morphologies in these SEM images are similar, consisting mostly of small grains. From this, it follows that the above-mentioned atmospheres did not significantly affect the morphologies of the samples.

SEM microphotographs of the surface morphology of B70Y30 after saturation at 600 °C for 600 h in a N2/H2O rich atmosphere and b CO2/H2O rich atmosphere
Conclusions
Combined DTA–TG–MS analysis exposed to CO2/H2O rich atmosphere shows that the chemical stability of the powders is proportional to the amount YDC15 in the samples. However, the porosity of the sintered materials also seems to increase along with the amount of YDC15 in the composite materials. Therefore, the higher the amount of YDC15 in a given bulk composite sample, the more similar the rate of reaction between BCY15 in the material and CO2 becomes to that of its respective powder at 600 °C. As a result, the slowest formation of BaCO3 takes place on the BCY15 sintered sample. However, all of the bulk samples managed to maintain cohesion despite the significant effects of CO2. DTA–TG–MS and XRD analysis also confirm that the effects of H2O at 600 °C on each of the materials are negligible. In conclusion, the results of this work suggest the possibility of producing BCY15–YDC15 composite samples that are better suited for operation in the temperature range 600–700 °C in CO2/H2O atmospheres than pure BCY15.
References
Iwahara H, Uchida H, Ono K, Ogaki K. Proton conduction in sintered oxides based on BaCeO3. J Electrochem Soc. 1988;135:529–33.
Hibino T, Hashimoto A, Suzuki M, Sano M. A solid oxide fuel cell using Y-doped BaCeO3 with Pd-loaded FeO anode and Ba0.5Pr0.5CoO3 cathode at low temperatures. J Electrochem Soc. 2002;149:A1503–8.
Shimura T, Tanaka H, Matsumoto H, Yogo T. Influence of the transition-metal doping on conductivity of a BaCeO3-based protonic conductor. Solid State Ionics. 2005;176:2945–50.
Suksamai W, Metcalfe IS. Measurement of proton and oxide ion fluxes in a working Y-doped BaCeO3 SOFC. Solid State Ionics. 2007;178:627–34.
Chiodelli G, Malavasi L, Tealdi C, Barison S, Battagliarin M, Doubova L, Fabrizio M, Mortaló C, Gerbasi R. Role of synthetic route on the transport properties of BaCe1−x Y x O3 proton conductor. J Alloy Compd. 2009;470:477–85.
Gopalan S, Virkar AV. Thermodynamic stabilities of SrCeO3 and BaCeO3 using a molten salt method and galvanic cells. J Electrochem Soc. 1993;140:1060–5.
Tanner CW, Virkar AV. Instability of BaCeO3 in H2O-containing atmospheres. J Electrochem Soc. 1996;143:1386–9.
Wu Z, Liu M. Stability of BaCe0.8Gd0.2O3 in a H2O-containing atmosphere at intermediate temperatures. J Electrochem Soc. 1997;144:2170–5.
Zakowsky N, Williamson S, Irvine JTS. Elaboration of CO2 tolerance limits of BaCe0.9Y0.1O3−δ electrolytes for fuel cells and other applications. Solid State Ionics. 2005;176:3019–26.
Ryu KH, Haile SM. Chemical stability and proton conductivity of doped BaCeO3–BaZrO3 solid solutions. Solid State Ionics. 1999;125:355–67.
Katahira K, Kohchi Y, Shimura T, Iwahara H. Protonic conduction in Zr-substituted BaCeO3. Solid State Ionics. 2000;138:91–8.
Lü J, Wang L, Fan L, Li Y, Dai L, Guo H. Chemical stability of doped BaCeO3–BaZrO3 solid solutions in different atmospheres. J Rare Earths. 2008;26:505–10.
Sawant P, Varma S, Wani BN, Bharadwaj SR. Synthesis, stability and conductivity of BaCe0.8−x Zr x Y0.2O3−δ as electrolyte for proton conducting SOFC. Int J Hydrogen Energy. 2012;37:3848–56.
Okiba T, Fujishiro F, Hashimoto T. Evaluation of kinetic stability against CO2 and conducting property of BaCe0.9−x Zr x Y0.1O3−δ . J Therm Anal Calorim. 2013;113:1269–74.
Pasierb P, Drożdż-Cieśla E, Gajerski R, Łabuś S, Komornicki S, Rękas M. Chemical stability of Ba(Ce1–x Ti x )1−y Y y O3 proton-conducting solid electrolytes. J Therm Anal Calorim. 2009;96:475–80.
Xie K, Yan R, Liu X. Stable BaCe0.7Ti0.1Y0.2O3−δ proton conductor for solid oxide fuel cells. J Alloys Compd. 2009;479:L40–2.
Bi L, Zhang S, Zhang L, Tao Z, Wang H, Liu W. Indium as an ideal functional dopant for a proton-conducting solid oxide fuel cell. Int J Hydrogen Energy. 2009;34:2421–5.
Caboche G, Hochepied J-F, Piccardo P, Przybylski K, Ruckdäschel R, Ardigó M-R, Fatome E, Chevalier S, Perron A, Combemale L, Palard M, Prazuch J, Brylewski T. Compatibility and reactivity between materials in an innovative dual membrane fuel-cell (IDEAL-cell) design. ECS Trans. 2009;25:763–72.
Thorel AS, Chesnaud A, Viviani M, Barbucci A, Presto S, Piccardo P, Ilhan Z, Vladikova D, Stoynov Z. IDEAL-cell, a high temperature innovative dual membrane fuel-cell. ECS Trans. 2009;25:753–62.
Presto S, Barbucci A, Viviani M, Ilhan Z, Ansar SA, Soysal D, Thorel A, Abreu J, Chesnaud A, Politova T, Przybylski K, Prazuch J, Brylewski T, Zhao Z, Vladikova D, Stoynov Z. IDEAL-Cell, an innovative dual membrane fuel-cell: fabrication and electrochemical testing of first prototypes. ECS Trans. 2009;25:773–82.
Gawel R, Viviani M, Przybylski K. Long-term chemical stability of BaCe0,85Y0,15O3−δ + Ce0,85Y0,15O2−δ composite samples at room temperature and at 873 K for use in dual PCFC-SOFC fuel cells. Arch Metall Mater. 2013;58:393–7.
Acknowledgements
The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement No 213389. The authors are also grateful to Marion Technologies S.A. for the synthesis and particle size distribution analysis of the initial BCY15 and YDC15 powders and Dr R. Gajerski for his help with the DTA-TG-MS analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gawel, R., Przybylski, K. & Viviani, M. Resistance of composite materials based on BaCeO3 against the corrosive effects of carbon dioxide and water vapour at intermediate fuel cell operating temperatures. J Therm Anal Calorim 116, 895–903 (2014). https://doi.org/10.1007/s10973-013-3562-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3562-9