
Abstract
Two series of new thermoplastic poly(thiourethane-urethane) elastomers (EPTURs), with different hard-segment content (40–60 wt%), were synthesized by a one-step melt polymerization from poly(oxytetramethylene) diol (PTMO) of \( \overline{M}_{\text{n}} \) = 1,000 g mol−1 or poly(hexamethylene carbonate) diol (PHCD) of \( \overline{M}_{\text{n}} \) = 860 g mol−1 as a soft segment, hexane-1,6-diyl diisocyanate and (methylenedi-1,4-phenylene)dimethanethiol as a chain extender at the NCO/(OH + SH) molar ratio of 1. The structures of all the EPTURs were examined by Fourier transform infrared spectroscopy (FTIR), atomic force microscopy and X-ray diffraction analysis. Their thermal behavior was investigated by means of differential scanning calorimetry and thermogravimetric analysis (TG). For the chosen polymers the gaseous products evolved during the decomposition process were analyzed by TG-FTIR. Moreover, physicochemical, adhesive and tensile properties as well as Shore A/D hardness were determined. The resulting high-molecular-mass EPTURs were stable up to 254–262 °C, as measured by the temperature of 1 % mass loss. They decomposed in three or four stages. The main decomposition products were carbonyl sulfide, isocyanate, carbon dioxide, aromatic hydrocarbons as well as aliphatic ethers and aldehydes (in PTMO series) and alcohols (in PHCD series). All the polymers showed partially crystalline structures, associated with crystallization of thiourethane hard segments. Their melting temperatures were in the range of 184–186 °C. The PTMO series EPTURs exhibited better low-temperature properties (glass-transition temperature in the range of −64 to −44 vs. −26 to −22 °C), but poorer tensile strengths (20–28 vs. 37–43 MPa). These EPTURs showed improved adhesive properties in comparison with their polyurethane analogs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfur-containing polymers, when considering their structure, make up a complex group of macromolecular compounds of varied and often remarkable features that determine their versatile applications. The presence of sulfur atoms in polymer structure, depending on the kind of functional group, can improve some important properties, such as mechanical, electrical and optical, and then adhesion to metals, resistance to heat, chemicals, radiation and bacteria, biocompatibility, and so on. Their more important functional groups are: sulfide, sulfonyl, sulfinyl, sulfo, as well as some organic groups containing sulfur atoms, such as thiourethane, thioester and thiocarbonate [1–4].
Among the polymers under consideration, great industrial significance has been gained by polysulfides, polysulfones, sulfopolymers and polysulfates. They are used mainly as high performance engineering plastics, elastomers with good resistance to environment and hydrocarbon fuels, chemically stable ion-exchange membranes in electromembrane processes, as well as materials in biomedical applications. Poly(thiourethane)s, poly(thioester)s and poly(thiocarbonate)s have been synthesized and investigated mainly as potential plastics with high thermal and chemical resistance, as well as with high refractive index for optical applications (i.e., lenses and optical fibers) [1, 4–7].
The less studied group are elastomers containing thiourethane groups, i.e. poly(thiourethane)s and poly(thiourethane-urethane)s. The earliest papers only present the results of the thermal properties of poly(thiourethane) elastomers based on oligobutadiene dithiol, decane-1,10-dithiol and 4,4′-diphenylmethane diisocyanate [8] and the investigation on hardness and tensile properties of rubbery poly(thiourethane)s synthesized from a few glycol bisthioglycolates, 2,4-tolylene diisocyanate or 4,4′-diphenylmethane diisocyanate and castor oil tristhioglycolate as a crosslinking agent. The latter showed poor tensile properties [9].
In the last decade only a few articles containing comprehensive studies on thermoplastic poly(thiourethane) elastomers [10] and poly(thiourethane-urethane) elastomers (EPTURs) [11–14], as well as crosslinked ones [15], have been published.
Shin et al. [10] have reported new thermoplastic poly(thiourethane) elastomers based on newly obtained oligo(thioether-ester) dithiol, hexane-1,6-dithiol as a chain extender, and aliphatic and aromatic diisocyanates. They have concentrated on examining the effect of the length of hard and soft segment, the ratio of these segments, and chemical structure of the hard segment on microphase separation and thermal and mechanical properties. The polymers were characterized by glass-transition temperature (T g) ranging from −57 to −23 °C and tensile strength and elongation at break up to 30 MPa and 1,000 %, respectively.
Gorna et al. [11, 12] have synthesized biodegradable thermoplastic EPTURs with the potential for applications in medical implants from biologically active chain extender, i.e. 2,2′-oxydiethanethiol, biodegradable poly(ε-caprolactone) diol of \( \overline{M}_{\text{n}} \) = 2,000 g mol−1, and hexane-1,6-diyl diisocyanate (HDI) or isophorone diisocyanate. The polymer with HDI showed lower T g (−51 vs. −38 °C) and tensile strength (about 22 vs. 31 MPa) and higher elongation at break (920 vs. 620 %). These polymers had molecular and mechanical properties comparable to those of commercial materials.
Thermoplastic EPTURs based on 2,2′-oxydiethanethiol, HDI and poly(ε-caprolactone) diol were also studied by Eglin et al. [13] as biodegradable and biocompatible materials in potential use in tissue engineering for cartilage and bone repairs.
We have taken up studies on the synthesis and characterization of new thermoplastic EPTURs derived from aliphatic–aromatic dithiol chain extenders, in contrast to aliphatic dithiols with acceptable odor, and various commercial oligomer diols and diisocyanates. In our previous paper [14], we describe two series of polymers (with hard-segment content of 22–69 wt%) obtained by melt polymerization from bis[4-(sulfanylmethyl)phenyl]methanone, HDI, and poly(oxytetramethylene) diol (PTMO) of \( \overline{M}_{\text{n}} \) = 1,000 g mol−1 or poly(hexamethylene carbonate) diol (PHCD) of \( \overline{M}_{\text{n}} \) = 860 g mol−1 as a soft segment. The PHCD yielded elastomers with significantly higher tensile strength (up to 33.0 vs. up to 21.4 MPa). Unfortunately, the PHCD-based polymers showed higher T g (down to −30 °C vs. down to −68 °C), less advantageous for elastomers. In both series, the polymers with high hard-segment content showed semicrystalline structures while the others remained amorphous.
Herein, we present related EPTURs obtained by using diphenylmethane-derivative chain extender, i.e. (methylenedi-1,4-phenylene)dimethanethiol (DT), which is easier to obtain and due to its lower melting temperature (T m) more suitable for the synthesis of polyurethane elastomers by melt polymerization with aliphatic diisocyanates. Application of this chain extender that introduces diphenylmethane units with active methylene groups into polymer structure facilitates the cross-linking of these polymers by peroxide and thus improves their mechanical strength properties [16]. The present study concentrates on defining the structure and thermal properties of EPTURs. Moreover, basic physicochemical, mechanical and adhesive properties were determined.
Additionally, it includes a characterization of the newly obtained poly(thiourethane) (PTUR) based on DT and HDI, building the thiourethane hard segment in these EPTURs.
In order to facilitate the interpretation of the results, syntheses of polyurethanes not containing chain extender DT (PTMO + HDI and PHCD + HDI) were also carried out, and their thermal and mechanical properties determined.
Experimental
Materials
DT (m.p. = 81–82 °C after recrystallization from ethanol) was obtained from suitable bis(chloromethyl)–compound through the thiouronic salt [17]. PTMO of \( \overline{M}_{\text{n}} \) = 1,000 g mol−1 and PHCD of \( \overline{M}_{\text{n}} \) = 860 g mol−1 were purchased from Sigma-Aldrich (St. Louis, USA). Before being used, the PTMO and PHCD were heated at 90 °C in vacuo for 10 h, while HDI and dibutyltin dilaurate (DBTDL) from Merck–Schuchardt (Hohenbrunn, Germany) were used as received. The polymerization solvent, N,N-dimethylformamide, with water content of less than 0.02 % was purchased from Merck (Darmstadt, Germany) and used as received. The solvent was stored over activated 3–4 Å molecular sieves.
Measurements
Reduced viscosities (η reds, dL g−1) of 0.5 % polymer solution in a phenol (Ph)/1,1,2,2–tetrachloroethane (TChE) (Ph/TChE) mixture with a mass ratio of 1:3 were measured in an Ubbelohde viscometer (Poland) at 25 °C.
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were obtained with a Nicolet 8700 FTIR spectrometer (Thermo Scientific, USA) using thin films.
Elemental analysis was performed with a Perkin–Elmer CHN 2400 analyzer (USA).
Thermogravimetric analysis (TG) was carried out with a Netzsch STA 449 F1 Jupiter thermal analyzer (Germany) in the range of 40–800 °C in helium (flow = 20 mL min−1), at the heating rate of 10 °C min−1. Sample masses about 10 mg were used. The composition of the gas evolved during the decomposition process was analyzed by a Bruker Tensor 27 FTIR spectrometer (Germany) coupled online to a Netzsch STA instrument by Teflon transfer line with 2 mm diameter heated to 200 °C. The FTIR spectra were recorded in the spectral range of 600–4,000 cm−1 with 16 scans per spectrum at 4 cm−1 resolution.
Differential scanning calorimetry (DSC) thermograms were obtained with a Netzsch 204 calorimeter (Germany) in the range of −100 to 200–250 °C. The reported transitions were taken from first and second heating scans. The scans were performed at the heating/cooling rate of 10 °C min−1 under nitrogen atmosphere (flow = 30 mL min−1). Sample masses of about 10 mg were used. T gs for the polymer samples were taken as the inflection point on the curves of the heat-capacity changes. T ms were read at endothermic-peak maxima.
X-ray diffraction (XRD) measurements were performed using a DRON-3 X-ray apparatus (Russia) with a copper tube and nickel filter. The XRD patterns of the investigated samples were obtained by measuring the number of impulses within a given angle over 6 s. The measurements were taken every 0.04°. The XRD patterns were analyzed by WAXSFIT computer program [18]. The program resolves a diffraction curve on diffraction peaks and amorphous halo which allows to estimate the degree of crystallinity. As crystalline peaks are assumed those whose full width at half-maximum (FWHM) is contained in the range of 0.7–1.4°.
Atomic force microscopy (AFM) was carried out on a Nanoscope V (VEECO, USA) microscope, in tapping mode in air. The phase data were recorded simultaneously. Silicon probes that were used (NSG30, NT-MDT, Russia) had a nominal spring constant of 20–100 N m−1. In order to compare the structure of all investigated samples, the imaging parameters were kept constant. Medium tapping technique was used to obtain phase images at 1 μm scan size. The specimens used were the cuttings from crude polymers after one-month conditioning at room temperature.
The hardness of the EPTURs was measured by the Shore A/D method on a Zwick 7206/H04 hardness tester (Germany) at 23 °C; and values were taken after 15 s.
Tensile testing was performed on a Zwick/Roell Z010 (Germany) tensile-testing machine according to Polish Standard PN-81/C-89034 (EN ISO Standard 527-1:1996 and 527-2:1996) at the speed of 100 mm min−1 at 23 °C; tensile test pieces 1 mm thick and 6 mm wide (for the section measured) were cut from the pressed sheet.
Press moulding was done with a Carver hydraulic press (USA) at 120–160 °C under 10–30 MPa pressure.
The single lap shear strength of the polymers to copper plate, 100 × 25 × 1.5 mm3, was measured in accordance with Polish Standard PN-EN 1465:2009 by using a Zwick/Roell Z010 (Germany). The adhesive joint, 12.5 × 25 × 0.2 mm3, was prepared by pressing the polymer between the ends of two copper plates at 135–155 °C (prepared according to PN-EN-13887:2005), and then leaving them under a pressure of 30 MPa to cool to room temperature. Next, the plates were fixed by tensile-testing machine clips and underwent tensile testing, the speed of 2 mm min−1 at 23 °C.
Polymer synthesis
EPTURs
The EPTURs, with the hard-segment content of ~40, 50 and 60 wt% were prepared by a one-step melt polymerization process from HDI, DT and PTMO or PHCD at the NCO/(OH + SH) molar ratio of 1.
The general procedure for the synthesis of the EPTURs by this method was as follows. DT and PTMO or DT and PHCD (0.01 mol together) and HDI (0.01 mol) were heated with stirring under dry nitrogen to 90 °C in an oil bath. A catalytic amount of DBTDL was added to the formed clear melt and polymerization rapidly began at vigorous stirring. The reaction temperature was gradually raised to 130 °C and the formed white rubber-like product was additionally heated at this temperature for 2 h.
The same conditions were also applied to the syntheses of polyurethanes obtained without using a chain extender (PTMO + HDI and PHCD + HDI).
FTIR (cm−1) of the PTMO–based EPTURs: 1,660–1,659 and 1,636–1,633 (nonbonded and H–bonded C=O stretching of the thiourethane group); 1,720–1,717 and 1,687–1,681 (nonbonded and H–bonded C=O stretching of the urethane group), 1525–1,523 and 1,513–1,511 (N–H bending of the urethane and thiourethane group); 3,310–3,275 (N–H stretching of the (thio)urethane group); 1,106–1,104 (C–O stretching of ether group); 1,478–1,462 (benzene ring); 2,936–2,931 and 2,855–2,854 (asymmetric and symmetric C–H stretching of CH2).
FTIR (cm−1) of the PHCD–based EPTURs: 1,660–1,659 and 1,637–1,633 (nonbonded and H–bonded C=O stretching of the thiourethane group); 1,740–1,738 (nonbonded C=O stretching of the carbonate group), ~1,715 (shoulder) (nonbonded C=O stretching of the urethane group and H-bonded C=O stretching of carbonate group) and 1,681–1,680 (H–bonded C=O stretching of the urethane group), 1523–1,520 and 1,512–1,511 (N–H bending of the urethane and thiourethane group); 3,313–3,276 (N–H stretching of the (thio)urethane group); 1,247–1,245 (C–O stretching of the carbonate group); 1,465–1,462 (C–C stretching of benzene ring); 2,934–2,931 and 2,860–2,858 (asymmetric and symmetric C–H stretching of CH2).
PTUR
PTUR was prepared by solution polymerization of an equimolar amount of DT and HDI (DMF, conc. ~20 wt%) which was carried out under dry nitrogen for 4 h at 85–90 °C in the presence of a catalytic amount of DBTDL. The polymer precipitated and then washed with methanol was dried at 80 °C in vacuo to constant mass. Yield 94 %.
FTIR (cm−1): 1,657 and 1,633 (nonbonded and H-bonded C=O stretching), 1510 (N–H bending) and 3,281 (N–H stretching) of the thiourethane group; 3,024 (C–H stretching) and 1,477 (C–C stretching) of benzene ring; 815 (C–H bending of p-disubstituted benzene ring); 2,931 and 2,856 (asymmetric and symmetric C–H stretching of CH2).
Elem. Anal.: Calcd. For C23H28O2N2S2: C, 64.45 %; H, 6.58 %; N, 6.54 %. Found: C, 64.08 %; H, 6.70 %; N, 6.69 %.
Results and discussion
Polymer synthesis and characterization
The new thermoplastic PTUR, synthesized by a step-growth polymerization of aliphatic–aromatic dithiol DT with HDI, was white solid, showing high resistance to organic solvents. It was insoluble in common organic solvents, such as N,N-dimethylformamide, tetrahydrofuran, dimethyl sulfoxide, N,N-dimethylacetamide, N-methyl-2-pyrrolidone, TChE as well as in a Ph/TChE mixture.
The related thermoplastic EPTURs were prepared in one-step process, according to Scheme 1, from DT and HDI, which formed thiourethane hard segments, and PTMO or PHCD building soft segments.

Synthesis of EPTURs
The EPTURs were white materials, which similarly as PTUR showed high resistance to organic solvents. They were completely or partially soluble only in a Ph/TChE mixture. The η red values determined for the EPTURs, ranging from 2.10 to 4.04 dL g−1, point to their high molecular mass. Table 1 gives designations and η red values for the EPTURs.
The chemical structures of the polymers were confirmed by elemental analysis and/or ATR-FTIR.
The ATR-FTIR spectrum of the PTUR (see Fig. 1) shows significant absorption of the thiourethane group. There are two absorption bands of the carbonyl group at 1,633 and 1,657 cm−1 (much less intensive) corresponding to stretching vibration of H-bonded and nonbonded C=O group, and bands at 3,281 and 1,510 cm−1 corresponding to N–H stretching and N–H bending vibrations. The spectra of the EPTURs exhibit several carbonyl bands, owing to the presence of thiourethane and urethane linkages in polymer structure, and in the case of the EPTURs derived from PHCD, also carbonate linkages. From the comparison of the spectra received for PTUR and EPTURs it follows that bands at 1,637–1,633 and 1,660–1,659 cm−1 are related to the H-bonded and nonbonded C=O stretching of the thiourethane group, and those at 1,687–1,680 and 1,720–1,715 cm−1 can be ascribed to H-bonded and nonbonded C=O stretching of the urethane group. In the spectra of PHCD-based EPTURs the band at about 1,715 cm−1 can also be ascribed to H-bonded C=O stretching of the carbonate group, whilst the band at 1,740–1,738 cm−1 originates from nonbonded C=O stretching of the carbonate group [19]. Moreover, in all spectra one band at 3,313–3,275 cm−1 connected with N–H stretching vibrations of thiourethane and urethane groups can also be observed, as well as two weakly separated bands at 1,513–1,511 and 1,525–1,523 cm−1 which can be associated with N–H bending vibrations of thiourethane and urethane group, respectively. Figure 1, for example, shows the spectra of polymers A-50 and B-50.

ATR-FTIR spectra of the PTUR and EPTURs A-50 and B-50
Thermal properties
The thermal behaviour of the PTUR and all EPTURs was investigated by means of DSC and TG. The numerical DSC and TG data obtained are summarized in Tables 2 and 3, respectively. The DSC curves of all the polymers are presented in Fig. 2, while thermogravimetric (TG) and differential TG (DTG) curves of the selected polymers are given in Figs. 3 and 4. For the purpose of comparison Tables 2 and 3 and Figs. 2 and 3 also contain data received for polyurethanes obtained without using a chain extender (PTMO + HDI and PHCD + HDI) as well as for polymer diols used as soft segments (PTMO and PHCD).

DSC curves of polymers: PTUR, PTMO-based EPTURs, PTMO + HDI and PTMO

DSC curves of polymers: PTUR, PHCD-based EPTURs, PHCD + HDI and PHCD

DTG and TG curves of the PTUR and EPTURs A-50 and B-50
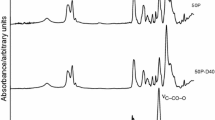
Moreover, for a better explanation of the process of decomposition of polymers through identification of the resulting volatile products, the TG coupled with Fourier transform infrared spectroscopy (TG-FTIR) was applied. The selected polymers were analyzed, i.e. PTUR and EPTURs A-50 and B-50. Figure 5 shows the FTIR spectra of decomposition products of polymers at the maximum rate of mass loss.

FTIR spectra of volatile products obtained at the maximum rate of mass loss of the thermal decomposition of the PTUR (for the first stage) and EPTURs A-50 (for the first, second and third stages) and B-50 (for the third stage)
DSC
The DSC curves of the EPTURs (after one-month conditioning at room temperature) from the first heating scans have a similar shape, and show distinct (with the exception of polymer A-60) glass transition of the soft segment in the range of −64 to −44 °C (from PTMO) and −26 to −22 °C (from PHCD), and one relatively sharp endothermic peak at 184–186 °C associated with the melting of thiourethane hard segments. High T m of those hard segments, close to that of PTUR and independent of their content, as well as high heat of melting (ΔH) values, point to the forming of well ordered thiourethane hard segment domains. Indistinct transitions, observed in the range of ~40–100 °C, may be connected both with the melting of low ordered hard segments and glass transition of amorphous portion of hard segments.
In contrast, the DSC curves from the second heating scans do not reveal crystallization on cooling from the melt at the applied cooling rate (10 °C/min) combined with absence of a long period of polymer conditioning at room temperature. And so the curves of the PTMO series EPTURs exhibit glass transition with high ΔC p values typical of amorphous polymers (−51 to −23 °C) and exothermic peaks at 46–86 °C resulting from “cold” crystallization of the thiourethane hard segments followed by endothermic peaks at 152–166 °C that correspond to their melting. It needs stressing that the enthalpy change for crystallization peaks and melting peaks are very similar; this also confirms that crystalline phase on cooling did not take place. In the case of the PHCD series EPTURs on the DSC curves only glass transition (−13 to 5 °C) is observed, this showing their lesser ability to crystallize on heating in comparison with the EPTURs of PTMO series. An increase in the mutual miscibility of the soft and hard segments during the first heating caused a shift of the glass transition to higher temperatures in both series of the EPTURs.
For both series of the EPTURs in both heating scans, a decrease in hard segment content resulted in a decrease in T g of the soft segment, suggesting an increase of the degree of microphase separation.
When the T g of the EPTURs and the respective soft segments were compared, it could be concluded that the PHCD-based EPTURs showed considerably lower degree of microphase separation. This may follow from lower molar mass of PHCD than PTMO soft segment, which with the same hard-segment content results in the PHCD-based polymers in higher fraction of HDI units not associated with DT, dispersed in the soft segment matrix [20]. It could also be the result of a stronger interaction of hard-segment thiourethane groups with carbonate groups of oligocarbonate chains than with oligoether chains of ether groups. On the other hand, the emergence of well separated thiourethane hard segment domains in polymers of both series indicates that the first parameter is crucial.
The newly obtained PHCD-based EPTURs showed significantly lower T g of the soft segment and hence better microphase separation than the analogous poly(thiourethane-urethane)s based on bis[4-(sulfanylmethyl)phenyl]methanone, already described by us [14]. It resulted from a smaller degree of compatibility between polycarbonate soft segments and hard segments formed from diphenylmethane-derivative compared with those from benzophenone-derivative chain extender. In the case of the PTMO-based polymers no significant effect of the kind of the chain extender used was observed on microphase separation.
TG
PTUR decomposition was a two-stage process, whereas the decomposition of the EPTURs occurred in three (for those from PTMO) or four stages (for those from PHCD). The DTG curve of the PTUR shows one large intense peak with maximum at 299 °C, connected with thiourethane bonds decomposition (as confirmed in TG-FTIR analysis, described below), and a significantly smaller and broader one at 417 °C, associated with the decomposition of the aromatic content in the polymer [21]. Comparison with the DTG curves of the PTUR and polymer diols suggests that in the case of the EPTURs the first stage (peak at 304–307 °C) can be ascribed to the decomposition of the thiourethane bonds and the second stage—the urethane bonds (weakly visible peak at 332–338 °C or as shoulder at 339–341 °C for the PTMO- or PHCD-based EPTURs, respectively). In the case of the PHCD-based EPTURs, the third stage (peak at 350–353 °C) is connected with the decomposition of the soft segment, whereas the forth stage (peak at 452 °C) is associated with the aromatic content in the polymer. For the PTMO-based EPTURs the forth stage is not observed, because the decomposition of this soft segment occurs in a temperature range similar to that of the aromatic content; hence the DTG curves show one broad peak instead of two (at 402–406 °C).
As can be seen from the TG data in Table 3, the EPTURs were characterized by better thermal stability as compared to the PTUR. The increase of hard-segment content in the EPTURs caused some reduction in T 5, T 10, and T 50. The polymers derived from PTMO generally showed somewhat higher T 1, T 5, T 10 and significantly much higher T 50 than those derived from PHCD. The reason for this is better stability of PTMO soft segment than that of PHCD soft segment.
TG-FTIR
The FTIR spectrum recorded during the first decomposition stage (299 °C) for the PTUR (Fig. 5) exhibits a very strong double absorption band at 2,071 and 2,047 cm−1, characteristic of asymmetric and symmetric C=O stretching vibration, and a weak band at 867 cm−1, attributed to C–S stretching vibration, in carbonyl sulfide [22–26]. This provides evidence that the decomposition of the PTUR occurred with the elimination of carbonyl sulfide, according to the following reaction:
On the other hand, the presence of a band at 2,280 cm−1 typical of asymmetric N=C=O stretching indicates another mechanism, attached to the dissociation of a thiourethane linkage to an aliphatic isocyanate and an aliphatic–aromatic thiol:
The appearance of bands at 2,359–2,309 and 669 cm−1 points to the formation of carbon dioxide probably as the result of partial carbodiimidization of isocyanate.
Moreover, the spectrum shows bands originating from aromatic hydrocarbons (toluene and xylene), i.e. at 3,105–3,082 cm−1 (C–H stretching vibration of benzene ring), at 1,507 cm−1 (C=C stretching vibration of benzene ring), at 1,064–1,033 cm−1 (C–H in-plane deformation vibration of monosubstituted benzene) and at 844 cm−1 (C–H bending vibration of p-disubstituted benzene derivatives). The evolution of these hydrocarbons results from the decomposition of the formed thiol and secondary amine. Bands at 1,541 and 1,523 cm−1 are a sign of the presence of carbon disulfide (asymmetric and symmetric C=S stretching vibration) [22–26], which can be generated in the reaction of carbonyl sulfide with HS radical derived from the thiol decomposition. The existence of isocyanate and hydrocarbons confirm bands at 2,929–2,890 cm−1 associated with asymmetric and symmetric C–H stretching vibration of methylene and methyl groups and a band at 1,374 cm−1 related to symmetric CH3-bending vibration from methyl derivatives of benzene and symmetric N=C=O stretching vibration.
The intensity of bands originating from carbonyl sulfide and those from isocyanate and carbon dioxide shows that the PTUR decomposition mainly occurred as proposed in reaction 1 above.
In the case of EPTUR A-50 (see Fig. 5), the first stage (306 °C) was characterized by the decomposition of the thiourethane hard segment, which is confirmed by evolution of the same products as for the PTUR (carbonyl sulfide, isocyanate, carbon dioxide, aromatic hydrocarbons, carbon disulfide). In the spectra recorded during the second stage (336 °C) bands originating from carbonyl sulfide (but of clearly lower intensity), isocyante and carbon dioxide (of slightly higher intensity) are still observed. The two former bands do not appear in the spectrum of the third stage. This fact means that in the second stage there was the termination of the decomposition of thiourethane linkages with simultaneous decomposition of urethane linkages. Because of the lack of bands characteristic of amine groups, it is reasonable to assume that urethane linkages decomposed as a result of dissociation to isocyanate (a volatile product) and polymeric alcohol, i.e. PTMO [27]. Thus, at the third stage (406 °C), mainly the decomposition of the PTMO soft segment took place, and the volatile products of its decomposition were aliphatic ethers, aldehydes and carbon monoxide. The same compounds have been identified by other authors in segmented polyurethanes with the PTMO soft segment and conventional hard segments [28, 29]. The presence of aldehydes was indicated by bands at 2,820 and 2,717 cm−1 connected with C–H stretching vibration, at 1,748–1,734 cm−1 related to C=O stretching vibration and at 950 cm−1 associated with C–H bending vibration of aldehyde group, while the release of ethers was evidenced by a band at 1,122 cm−1 connected with C–O stretching vibration of the ether group. Aliphatic character of these compounds was confirmed by bands at 2,966–2,875 cm−1 attributed to C–H stretching vibration of methylene and methyl groups. The existence of carbon monoxide was detected as a band at 2,186 cm−1. Moreover, the spectrum exhibits bands at 2,359–2,310 and 669 cm−1 corresponding to carbon dioxide and bands at 1508, 1457, 1374 and 689 cm−1 pointing to the presence of aromatic hydrocarbons, which may be formed due to the decomposition of the nonvolatile products obtained at the first and second stages of decomposition.
In the case of EPTUR B-50, the FTIR spectra obtained at the first, second and third stages (305–350 °C) show the bands attributed to the decomposition products of thiourethane and urethane linkages, noticed earlier for polymer A-50. At the third stage (see Fig. 5) there were evident additional bands at 1,054 and 3,677–3,594 cm−1 connected with C–O stretching vibration and O–H stretching vibration from alcohols; also, there was a rise in the intensity of bands at 2939–2,876 and 2,359–2,310 cm−1. This is indicative of the fact that at this stage the decomposition of the PHCD soft segment took place, accompanied by the evolution of aliphatic alcohols and carbon dioxide.
XRD analysis
DSC analysis indicated a relatively high degree of ordering in the PTUR and EPTURs. To determine the kind of ordering in these polymers XRD studies were done. The analysis of the XRD patterns by the WAXSFIT program showed that all the polymers had partially crystalline structures. The degrees of crystallinity (calculated as the ratio of the sum of crystalline to the total sum of crystalline and amorphous peak areas), given in Table 1, increased with the increase of hard-segment content in a polymer. The diffraction data also showed that crystallinity phase existing in the EPTURs was formed within thiourethane hard segments. This is evidenced by the fact that in the plots of the EPTURs were present only the same crystalline peaks (at 2θ ~8.0, 19.4, 20.5, 22.6, 25.3°) as those observed in the PTUR plot. Figure 6 presents the plots received for the PTUR and selected EPTURs, while Table 4 gives equivalent numerical data. The results obtained by XRD and DSC methods show good correlation (see discussion in DSC subchapter).

XRD curves (points) of a PTUR, b EPTUR A-60 and c EPTUR B-60 resolved into crystalline and amorphous peaks (solid lines)
AFM
To confirm the existence of microphase-separated morphology of the EPTURs, indirectly deduced from DSC results, tapping mode AFM experiments were performed. Tapping mode AFM generates phase images with dark features corresponding to the regions of lower modulus (soft-segment-rich domains) and bright features corresponding to regions of higher modulus (hard-segment-rich domains). The obtained phase images of all the EPTURs are given in Fig. 7. These data exhibit heterogeneous bulk morphology in the polymers examined, although there are some differences in the appearance of the images obtained, both within each series of polymers, and between the PTMO and PHCD series.

AFM phase images of EPTURs based on PTMO and PHCD: a A-40, b A-50, c A-60, d B-40, e B-50, f B-60
In the PTMO series for EPTURs A-40 and A-50 there are observed bright, filament-like features upon a dark area, which are shorter and wider for the EPTUR with higher hard-segment content. These structures correspond to linearly linked hard-segment domains and show a tendency toward parallel orientation. On the image for EPTUR A-60 the area associated with hard-segment domains increases and the difference between soft and hard phases is less pronounced which points to greater degree of phase mixing.
For the EPTURs based on PHCD smaller diversity in the appearance of the obtained phase images is observed when compared with those based on PTMO. This may be due to higher stiffness of polycarbonate than polyether chains, and hence smaller difference in stiffness between soft and hard segment chains, and a greater degree of phase mixing. This correlates with the results of DSC analysis which showed T g values in PHCD series higher and almost the same for all polymers.
Mechanical properties
Shore A/D hardness and tensile properties were determined for all the EPTURs and polyurethanes obtained without using a chain extender (PTMO + HDI and PHCO + HDI). Pressing temperatures and numerical data are shown in Table 5.
As is evident from the data presented in Table 5, the PHCD-based EPTURs showed significantly higher tensile strength (36.8–42.6 vs. 20.4–28.0 MPa) than those based on PTMO, with smaller differences in elongation at break (278–532 vs. 203–654 %). Moreover, these former polymers were characterized by somewhat higher modulus of elasticity (32.2–83.9 vs. 30.9–72.0 MPa) and slightly higher hardness in relation to the latter ones.
In the PTMO series, the increase in hard-segment content resulted in a decreased tensile strength, may be attributed to the increasing degree of phase mixing. For the PHCD-based EPTURs showing greater degree of phase mixing this above parameter is less significant, and the decrease in the tensile strength was observed for the polymer with 60 wt% hard-segment content. Moreover, in both series of EPTURs as the hard-segment content increased hardness and modulus of elasticity increased, while elongation at break decreased.
All the EPTURs exhibited a much higher tensile strength as compared to the polyurethanes prepared without using a chain extender. Their tensile strength was also improved in relation to poly(thiourethane-urethane)s obtained from bis[4-(sulfanylmethyl)phenyl]methanone as a chain extender [14].
Adhesive properties
The effect of introduction of sulfur atoms to polyurethane chain was investigated by comparing the lap shear strengths to copper between the synthesized EPTURs and their oxygen analogs obtained from (methylenedi-1,4-phenylene)dimethanol as a chain extender. For these studies the polymers with hard-segment content of 50 wt% were chosen.
As seen from the data given in Fig. 8, partial replacement of urethane linkages by thiourethane linkages caused about 20 % increase of the adhesive strength on copper–polymer junction.

Lap shear strength of selected EPTURs and their oxygen analogs
Conclusions
High molecular mass EPTURs with hard-segment content of 40–60 wt% were prepared by one-step melt polymerization from HDI, (methylenedi-1,4-phenylene)dimethanethiol (DT) as a chain extender and PTMO or PHCD as soft segments. All the polymers showed partially crystalline structures, associated with crystallization of thiourethane hard segments. T ms of these hard segments were in the range of 184–186 °C. The use of PHCD produced polymers with significantly higher tensile strengths (36.8–42.6 vs. 20.4–28.0 MPa) and unfortunately higher T gs (−26 to −22 vs. −64 to −44 °C), which to some extent limits the possibility of applications of these polymers as elastomers within negative temperatures. The TG results showed that the kind of the polymer diol used had no significant influence on thermal stability of the EPTURs whose temperatures of 1, 5 and 10 % mass loss were similar. For both types of polymers the decomposition was a multistage process, in three stages for those from PTMO or four stages for those from PHCD. The decomposition of the hard segment was mostly accompanied by the evolution of carbonyl sulfide, isocyanate, carbon dioxide and aromatic hydrocarbons, the PTMO soft segment decomposed into aldehydes and aliphatic ethers, while the PHCD soft segment into carbon dioxide and alcohols. In both series of the EPTURs, decreased hard-segment content resulted in decreased degree of crystallinity, T g of the soft segment, hardness and modulus of elasticity, as well as in the increased tensile strength and elongation at break. The studies of adhesive properties undertaken by us showed that the introduction of sulfur atoms to polyurethane chain caused somewhat improved adhesive strength of the copper-polymer interface.
References
Kultys A. Sulfur-containing polymers. In Mark HF, editor. Encyclopedia of polymers science and technology, vol 4. Hoboken: Wiley; 2003. p. 336–93.
Kricheldorf HR, Schwarz G. Poly(thioester)s. J Macromol Sci A. 2007;44:625–49.
Hirano H, Kadota J, Agari Y, Harada T, Tanaka M, Hasegawa K. Linear polymers with sulfur in the main chain. IV. Synthesis of thermotropic liquid-crystalline polythioesters based on 4,40-biphenyldithiol with excellent adhesive properties. Polym Eng Sci. 2007;47:262–9.
Tyagi M, Suri G, Chhabra P, Seshadri G, Malik A, Aggarwal S, Khandal RK. Novel way of making high refractive index plastics; metal containing polymers for optical applications. E-Polymers 2009;100.
Vietti D. Polysulfides. In: Mark HF, editor. Encyclopedia of polymers science and technology, vol 3. Hoboken: Wiley; 2003. p. 732–44.
El-Hibri MJ, Weineberg SA. Polysulfones. In Mark HF, editor. Encyclopedia of polymers science and technology, vol 4. Hoboken: Wiley; 2003. p. 1–25.
Li Q, Zhou H, Wicks DA, Hoyle CE, Magers DH, McAlexander HR. Comparison of small molecule and polymeric urethanes, thiourethanes, and dithiourethanes: hydrogen bonding and thermal, physical, and mechanical properties. Macromolecules. 2009;42:1824–33.
Lipatov YS, Muller S, Privalko VP, Singer H. Untersuchung der thermischen eigenschaften von polythiourethan-elastomeren auf der basis von oligobutadien. Angew Makromol Chem. 1979;82:79–87.
Ghatge ND, Murthy RAN. Polythiourethane polymers using castor oil tristhioglycolate as crosslinking agent. J Appl Polym Sci. 1982;27:1557–63.
Shin J, Matsushima H, Chan JW, Hoyle CE. Segmented polythiourethane elastomers through sequential thiol-ene and thiol-isocyanate reactions. Macromolecules. 2009;42:3294–301.
Gorna K, Polowinski S, Gogolewski S. Synthesis and characterization of biodegradable poly(ε-caprolactone urethane)s. I. Effect of the polyol molecular weight, catalyst, and chain extender on the molecular and physical characteristics. J Polym Sci A. 2002;40:156–70.
Gorna K, Gogolewski S. In vitro degradation of novel medical biodegradable aliphatic polyurethanes based on ε-caprolactone and Pluronics® with various hydrophilicities. Polym Degrad Stab. 2002;75:113–22.
Eglin D, Griffon S, Alini M. Thiol-containing degradable poly(thiourethane-urethane)s for tissue engineering. J Biomat Sci. 2010;21:477–91.
Kultys A, Rogulska M, Pikus S. The synthesis and characterization of new thermoplastic poly(thiourethane-urethane)s. J Polym Sci A. 2008;46:1770–82.
Novakov IA, Chalykh AE, Nistratov AV, Reznikova OA, Matveev VV, Budylin NY, Pylnov DV. Study of the structure and curing of thiourethane elastomers based on oligomer compositions. Polym Sci B. 2012;54:240–6.
Wirpsza Z. Polyurethanes: chemistry, technology and applications. New York: Ellis Horwood; 1993.
Podkoscielny W, Podgorski M. Linear polythioesters. 26. Products of interfacial polycondenation of bis(4-mercaptomethylphenyl)methane with some aliphatic and isomeric phthaloyl dichlorides. Angew Makromol Chem. 1996;241:135–48.
Rabiej M, Rabiej S. Analysis of X-ray diffraction pattern of polymers by means of WAXSFIT computer program (in Polish). Poland: ATM; 2006.
Hernandez R, Weksler J, Padsalgikar A, Choi T, Angelo E, Lin JS, Xu LC, Siedlecki CA, Runt J. A comparison of phase organization of model segmented polyurethanes with different intersegment compatibilities. Macromolecules. 2008;41:9767–76.
Martin DJ, Meijs GF, Renwick GM, McCarthy SJ, Gunatillake PA. The effect of average soft segment length on morphology and properties of a series of polyurethane elastomers. 1. Characterization of the series. J Appl Polym Sci. 1996;62:1377–86.
Xu Y, Petrovic Z, Das S, Wilkes GL. Morphology and properties of thermoplastic polyurethanes with dangling chains in ricinoleate-based soft segments. Polymer. 2008;49:4248–58.
Dunn JG, Chamberlain AC, Fisher NG, Avraamides J. The influence of activated carbon on the thermal decomposition of sodium ethyl xanthate. J Therm Anal Calorim. 1997;49:1399–408.
Fisher NG, Dunn JG. Analysis of a complex gaseous mixture by TG-MS and TG-FTIR. J Therm Anal Calorim. 1999;56:43–9.
Madarász J, Pokol G. Comparative evolved gas analyses on thermal degradation of thiourea by coupled TG-FTIR and TG/DTA-MS instruments. J Therm Anal Calorim. 2007;88:329–36.
Otto K, Bombicz P, Madara’sz J, Oja Acik I, Krunks M, Pokol G. Structure and evolved gas analyses (TG/DTA-MS and TG-FTIR) of mer-trichlorotris(thiourea)-indium(III), a precursor for indium sulfide thin films. J Therm Anal Calorim. 2011;105:83–91.
Ahamad T, Alshehri SM. Thermal degradation and evolved gas analysis of thioureaformaldehyde resin (TFR) during pyrolysis and combustion. J Therm Anal Calorim. 2012;109:1039–47.
Shufen L, Zhi J, Kaijun Y, Shuqin Y, Chow WK. Studies on the thermal behavior of polyurethanes. Polym-Plast Technol. 2006;45:95–108.
Herrera M, Matuschek G, Kettrup A. Thermal degradation of thermoplastic polyurethane elastomers (TPU) based on MDI. Polym Degrad Stab. 2002;78:323–31.
Cervantes-Uc JM, Moo Espinosa JI, Cauich-Rodrıguez JV, Avila-Ortega A, Vazquez-Torres H, Marcos-Fernandez A, San Roman J. TGA/FTIR studies of segmented aliphatic polyurethanes and their nanocomposities prepared with commercial montmorillonites. Polym Degrad Stab. 2009;94:1666–77.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Rogulska, M., Kultys, A. & Olszewska, E. New thermoplastic poly(thiourethane-urethane) elastomers based on hexane-1,6-diyl diisocyanate (HDI). J Therm Anal Calorim 114, 903–916 (2013). https://doi.org/10.1007/s10973-013-3007-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3007-5