
Abstract
Three substituent derivatives of glycerol dimethacrylate (GDMA) i.e., acetyloxypropyl dimethacrylate (Acet-GDMA), acryloyloxy-hydroxypropyl methacrylate (GMMA), and acetyloxy-acryloyloxypropyl methacrylate (Acet-GMMA) as well as GDMA were prepared and UV-homopolymerized in the presence of 2,2-dimethoxy-2-phenyloacetophenone. The obtained homopolymers were subjected to dynamic mechanical studies and the deflections versus temperature measurements (HDT) were performed. The content of unreacted double bonds, glass transition temperatures (T g) as well as degree of inhomogeneity have been examined in all studied systems. Also, expansivity was measured in the function of temperature and the effect of a substituent group on the network properties was investigated. The results show that the hydroxyl-containing networks are characterized by much lower T g and higher degree of heterogeneity than those devoid of hydroxyls. When homopolymerized, hydroxyl-free dimethacrylates disclosed untypical deflection pattern which suggests that at higher temperatures the decrease in stiffness is compensated or even overrated by the increase in the specific volume. Expansivity measurements revealed twice as large volume increase of the acetylated polymers as opposed to their hydroxyl-containing analogs. Following these findings, a mechanism of thermally induced polymer “swelling” was proposed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Photo-, and thermally initiated polymerization of multifunctional (meth)acrylates produces densely crosslinked polymers with high thermal stability, mechanical strength and chemical resistance. Because of good thermo-mechanical properties, crosslinked (meth)acrylates have found use in a wide variety of applications. They are successfully employed in coatings for flooring and furniture, dental restorative materials, optical fibers, hard and soft contact lenses, and photolithography [1–6].
Crosslinked polymers owe their exceptional properties to the intricacy of the network. Over the past years extensive studies have been utilized to evaluate tri-dimensional network formation. It has been established that during crosslinking polymerization of multifunctional monomers, networks of high degree of inhomogeneity are formed [7–12]. This is because the polymerization with crosslinking exhibits specific reaction behaviors, including prominent autoacceleration, autodeceleration, and reaction-diffusion controlled termination [13–15]. In consequence, functional groups differ in reactivity, and the overall conversion is limited due to hindered mobility of reactive groups [16]. Attempts at increasing network homogeneity have been made and these include the atom transfer radical polymerization [17], controlled/living radical polymerization [18, 19], or recently applied thiol-ene-methacrylate systems with step growth polymerization mechanism [20].
As in any other system, the properties (structure) of the network that evolve during crosslinking polymerization depend on the structures of monomers that are polymerized. Specifically, their type and functionality, length and flexibility of spacer groups, presence/lack of heteroatoms or cyclic structures or aliphatic/aromatic rings have been widely considered [21–28]. It is now well-known that the increasing monomer functionality increases crosslinking density, but it also increases network heterogeneity and decreases conversion [21]. Monomers with long and flexible spacer chains between unsaturations tend to produce loose networks with high degrees of conversion [22] whereas polymerization of stiff-structured monomers results in rigid networks and low double bond conversions [23]. On the other hand, the effect of indifferent pendant group on the evolution of properties of highly crosslinked networks with the increase of temperature has not been well studied.
The present study aims to investigate the effects the side pendant groups exert on the network properties and the volume–temperature dependence. Better understanding of how crosslinked polymers with different pendant functionalities undergo structural and property changes in response to external stimuli (temperature) should enable a more efficient use of such materials in many applications. So as to achieve this goal, low molecular weight di(meth)acrylate monomers of similar chemical design with different pendant side groups were synthesized and homopolymerized. The monomers were derived from glycerol dimethacrylate and its mono-acrylate/mono-methacrylate analog by acetylation of their hydroxyl groups. The features like relatively low molecular weight and low viscosity make them very effective in crosslinking polymerization. Their highly crosslinked networks were studied by DMA and HDT. Also, the monomers were thermally cured and their polymerization conversion was evaluated by DSC.
Experimental
Materials
Glycidyl methacrylate (GMA, 97 %, from Sigma-Aldrich Chemie GmbH), tetraethylammonium bromide (analytically pure, from Merck KGaA Frankfurt), methacrylate and acrylate acids (99 %, from Merck KGaA Frankfurt), hydroquinone (analytically pure, from Merck KGaA Frankfurt), 2,2-dimethoxy-2-phenyloacetophenone (Irgacure 651, UV polymerization initiator, from Ciba Specialty Chemicals), benzoil peroxide (BPO, analytically pure, from Sigma-Aldrich Chemie GmbH)), sulfuric acid (98 %, from POCh Gliwice), acetic anhydride (analytically pure, from POCh Gliwice), diethyl ether (analytically pure, from POCh Gliwice), anhydrous magnesium sulfate (analytically pure, from POCh Gliwice), and sodium hydrogen carbonate (analytically pure, from POCh Gliwice) were used as received.
Synthesis of di(meth)acrylates
Glycerol dimethacrylate (a mixture of isomers) (GDMA) and its mono-acrylate analog (GMMA) were prepared by the reaction of glycidyl methacrylate with methacrylic and/or acrylic acid. The acid-epoxide addition was carried out in the presence of tetraethylammonium bromide. Both syntheses were stabilized by hydroquinone. The detailed procedures are described elsewhere [29]. The acetylated di(meth)acrylates, acetyloxypropyl dimethacrylate (Acet-GDMA) and acetyloxy-acryloyloxypropyl methacrylate (Acet-GMMA) were obtained by reacting acetic anhydride with GDMA and GMMA, respectively. The general procedure, described in detail for Acet-GDMA in the study [30], is as follows. A mixture of GDMA (0.438 m), acetic anhydride (0.446 m) and sulfuric acid (0.05 g) was kept at 80–85 °C for 3 h. After cooling, the mixture was dissolved in diethyl ether and washed repeatedly with distilled water, 5 wt% of aqueous NaHCO3, and distilled water again. After drying over anhydrous magnesium sulfate and filtration, the solvent was removed under reduced pressure at 50 °C.
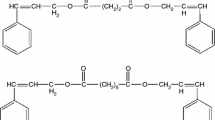
The purified products were obtained in the yield of 79 % for Acet-GDMA (94 g from the initial 100 g of GDMA) and 66 % for Acet-GMMA (79 g from the initial 100 g of GMMA). Molecular structures of the di(meth)acrylates are shown in Fig. 1. They have been confirmed by H NMR spectra and elemental analysis. GMMA: δ 1.9 (3H), 4.3 (4H), 4.4 (1H), 5.6, 6.2 (2H), 6.4, 5.8 (2H), 3.8 (2H), 6.1 (1H); Acet-GMMA: 1.9 (3H), 5.3 (1H), 4.4 (4H), 5.6, 6.2 (2H), 6.4, 5.8 (2H), 4.3 (2H), 2.1 (3H) 6.1 (1H).

Chemical structures of di(meth)acrylate monomers with their abbreviations
Elemental analysis of the obtained di(meth)acrylate esters is as follows.
The theoretical composition of GMMA: %C: 56.07; %H: 6.59; the determined composition: %C: 55.81; %H: 6.65.
The theoretical composition of Acet-GMMA: %C: 56.24; %H: 6.29; the determined composition: %C: 55.76; %H: 6.18.
Since the catalyst (tetraethylammonium bromide) is characterized by rather small selectivity, the monomers consist of mixtures of isomers. In the present study, this is confirmed by the presence of bands for protons of =CHOH groups (4.4 ppm) and 3.8 ppm bands for protons of –CH2OH groups. Likewise, in the NMR spectra of the acetylated di(meth)acrylates there are signals for protons of =CHOR groups (5.3 ppm) and bands for protons of –CH2OR groups (4.3 ppm). Based on the NMR analysis, it was evaluated that the isomers with primary hydroxyl groups constitute around 20–25 % of the total [30].
Curing procedure
The monomers were irradiated by an ultraviolet light (340–360 nm) in the presence of a photoinitiator (Irgacure 651) used in the amount of 1 wt%. Photopolymerization was carried out in glass molds and initiated with four TL20 W/05 SLV low-pressure mercury lamps at 20 °C. The compositions in the molds were irradiated from both sides for 10 min. The homopolymers subsequently underwent thermal post-curing at the temperatures above their tan δmax i.e., at 160 °C for 6 h. Long-term post-curing produces thermally stable networks with lasting properties [31]. Post-cured polymers could be repeatedly heated and cooled down and the polymerization should not recommence.
Research methods
1H-NMR spectra of raw methacrylate monomers were recorded at 20 °C on an FT-NMR Bruker Avance (Germany) spectrometer operating at the 1H resonance frequency of 300 MHz. Chemical shifts were referenced to tetramethyl silane serving as an internal standard.
Elementary analysis of the obtained methacrylate esters and substrates used in the syntheses was carried out using the Perkin Elmer CHN 2400 apparatus.
Viscosities of the methacrylate monomers were measured at different temperatures by means of a rotating spindle rheometer (Brookfield, model DV-III) using appropriate spindles and standard solutions. Viscosity was measured at various spindle speeds (5–150 rpm) and only readings obtained around 50 % torque were recorded and expressed as mPas.
The effect of temperature on viscoelastic properties of the obtained polymers in the area of linear dependence between stress and strain was determined using a DMA Q800 apparatus produced by TA (USA). Thermo-mechanical properties of the cured polymers were evaluated from storage modulus (E’), and tan δ curves obtained at constant frequency (10 Hz). Measurements for all samples were made in the temperature range 10–260/280 °C at a constant heating rate of 4 °C/min. Rectangular profiles of the sizes: a = 4 ± 0.2 mm, b = 10 ± 0.2 mm and c ≥ 35 mm were used in the measurements.
Thermal resistance under loading was evaluated using an HDT 3 VICAT apparatus produced by CEAST S.p.A (Italy). Specimens of (4 ± 0.2 mm) × (10 ± 0.2 mm) × (70+ mm) were submerged in an oil bath, and the oil bath heated from 20 to 200/300 °C with an initial soak time of 5 min at a heating rate of 2 °C/min. The heat distortion temperature was detected by a linear variable displacement transducer as the temperature at which a 2 % deflection under a load of 1.8 MPa occurred. The reported HDT was an average of three measurements.
Expansivity measurements were conducted at atmospheric pressure on an HDT 3 VICAT apparatus (Scheme 1). The sample, a rectangular profile of ~(10 ± 0.2 mm) × (10 ± 0.2 mm) × (4 ± 0.2 mm) is placed on a steel table. A movable steel rod with an adjustable flat end is pressed to the upper end of the sample. The other end of the rod is connected with a linear variable displacement transducer which detects displacements with the precision of 1 μm. The steel rod exerts an additional pressure of 5 hPa on the sample which is neglected since it corresponds to the daily fluctuations of atmospheric pressure. During the experiments, specimens are submerged in an oil bath, and the oil bath is heated from 20 to 220 °C with an initial soak time of 5 min at a heating rate of 2 °C/min. The heat transfer from the silicone oil to the sample is reckoned to be satisfactory due to small size of the samples and the continuous circulation of the oil. The thermocouple that indicates the sample temperature is placed a few millimeters apart from it in the silicone oil. The rate of temperature change should be also satisfactory because the measurements with the slower rate of temperature change (1 °C/min) did not lead to significantly better or different results. Therefore, all measurements were made with the speed of 2 °C/min. Before the actual measurements, thermal expansion of the apparatus itself was evaluated by taking sample-free runs and the data for the samples were corrected accordingly.

Schematic view of the chamber of HDT/Vicat apparatus used for volume–temperature measurements
Differential scanning calorimetry (DSC) measurements were performed on a NETZSCH 204 apparatus at 10 °C/min. The calorimeter was calibrated with indium (ΔH fusion 28.6 J/g, melting point 156.6 °C) prior to use. An empty aluminum pan was used as a reference.
Results and discussion
Physical properties of di(meth)acrylates
Glycerol dimethacrylate and its derivatives are yellowish liquids with relatively low viscosities. The acetylated di(meth)acrylates have slightly lower viscosities than their hydroxyl-containing counterparts. This is attributed to the elimination of intermolecular hydrogen bonding owing to hydroxyl groups. Besides, the monomers have comparable densities and refractive indices. These are slightly influenced by acetylation which causes an increase in densities and decrease in refractive indices (Table 1).
DMA of the obtained polymers
The DMA scans along with glass transition temperatures are summarized in Figs. 2, 3 and Table 2.

Storage modulus versus temperature for homopolymerized di(meth)acrylates

Tangent δ versus temperature for homopolymerized di(meth)acrylates

Heat deflection versus temperature measurements for di(meth)acrylate homopolymers

Volumetric expansion in the function of temperature for the studied polymers
In Fig. 2 the storage moduli in the function of temperature for the obtained homopolymers are depicted. As can be seen from them, the modulus behavior is well captured and there is no indication of a continued reaction during the temperature scans. This means that the polymers are thermally stable and their networks could be heated without facilitating further crosslinking. Examining Fig. 2, it can be seen that polymers with hydroxyl groups attached to their networks exhibit higher storage moduli as compared to those with acetylated groups. This tendency is well maintained throughout the analysis. It is therefore possible that hydrogen bonding acting as an additional semi-bonding in the network increases its stiffness. Further, the rubber-like state of three of the networks studied is not attained in the temperature interval of the experiments. Due to their brittleness, the samples brake at the end of the temperature runs and the rubbery plateau cannot be achieved in the current experimental conditions.
As can be seen from the width of the tan δ curves (Fig. 3), it spreads over a very wide temperature range. This extended transition region could be a result of high degree of structural heterogeneity of the networks in which different types of relaxations are superposed. Usually, the broader the tan δ peak the more heterogeneous polymer with a wide distribution of relaxation times [8, 9]. Because the glass transition region is spread over a wide temperature range, it is difficult to provide a specific temperature as the glass transition temperature of the material. Therefore, similar to the previous reports, the T g was taken as the temperature at the peak of the tan δ curve in the glass transition region.
Examining the information in Fig. 3, it can be seen that homopolymers obtained from acetylated di(meth)acrylates exhibit symmetrical tan δ plots with only one maximum. On the other hand, the tan δ curves of hydroxyl-containing di(meth)acrylates are asymmetrical. In the case of the GDMA homopolymer, the peak comes out at around 80 °C but there is also a shoulder in the curve discernible at above 200 °C. A similar tendency can be observed in the tan δ curve of the GMMA homopolymer where a shoulder is visible at 80 °C and a broad peak comes out at higher temperatures. In DMA, the glass transition temperature is associated with the process of segmental relaxation (α) whereas β relaxation, which precedes α relaxation at lower temperatures, implies motions or/and interactions of side groups [32]. It appears that the GDMA homopolymer should have its T g far beyond the tan δ peak value which may be caused by strong interactions of hydroxyl groups and hydrogen bonding. Therefore, it is possible that the T g is marked by the shoulder in the vicinity of 200 °C. The actual value of T g for the GMMA homopolymer could be understated due to distinct hydrogen bonding too. Further, the symmetrical tan δ plots of the acetylated homopolymers do not necessarily mean single α relaxations. It is possible that the peaks cover α as well as β relaxation. This may be due to overlapping of both relaxations of which α is reduced in highly crosslinked network whereas β is intensified by ample amount of acetyl groups and pendant unsaturations. Similar tendency was observed in one of my previous studies [33]. Besides, the glass transition temperature could depend on the degree of structural heterogeneity [29].
Examining Fig. 3 further, it can be seen that the GDMA and GMMA homopolymers have significantly broader widths and lower values of the tan δ peaks than their acetylated analogs.
As was already mentioned, the heterogeneity of the network manifests itself as a wide distribution of relaxation times because there exists a very broad distribution of mobilities in the matrix. It can be qualitatively evaluated by examining the width of the tan δ peak. Regarding the results in Fig. 3, it is evident that hydroxyl-containing monomers produce networks with higher distribution of relaxation times than their acetylated counterparts.
It could be presumed that the quasi-network hydrogen bonded structure which probably forms during polymerization of hydroxyl-containing monomers increases the heterogeneity of the final polymer.
Interestingly, the polymers with high degree of structural heterogeneity may have their T g shifted towards lower values. When comparing the homopolymers of GDMA and Acet-GDMA, the latter has more homogeneous structure and higher T g value. It appears that high network heterogeneity as well as overlapping of both relaxations are plausible reasons for the decreased glass transition temperature.
HDT and expansivity measurements
In Fig. 4, the HDT curves of the di(meth)acrylate homopolymers are depicted. The values of HDT decrease in the order of poly-GDMA > poly-GMMA > poly-Acet-GDMA > poly-Acet-GMMA. The HDT plots for GDMA and GMMA homopolymers have typical shapes where deflection exponentially increases with temperature. Different shapes of HDT plots were obtained for the acetylated homopolymers.
Under a load of 1.8 MPa all polymer samples start to gradually deflect with the increase of temperature. In the first stage of a temperature increase, the largest displacement is observed for the GMMA homopolymer. At the temperatures above 100 °C, the acetylated polymers cease to deflect, the Acet-GDMA homopolymer even reverts its distortion to some extent. At the temperatures above 160–180 °C they deflect again until the final HDT values are achieved. In a temperature range from 100 to 180 °C, there can be observed an apparent increase in stiffness. However, as indicated by the DMA results all discussed polymers lose their modulus as the temperature increases over the entire range of analysis. Since the chemical reaction can be ruled out (the specimens have been thermally post-cured at 160 °C), a physical process must be involved in this anomalous behavior.
It appears that the observed tendencies can be partially explained by the thermal volumetric expansion of the studied networks. This physical process has to be significant since it withstands or even overcomes the static force of the 1.8 MPa load. In other words, the specific volume of the acetylated polymers seems to increase faster than they deflect. It might be the cause of the plateau as well as the reversal displacement seen in the HDT plots of Acet-GMMA and Acet-GDMA homopolymers.
For the isotropic samples volume changes are easily calculated from the change in one dimension [34]. In the present study, the specific volume V was calculated according to
with the V 0 specific volume at atmospheric pressure and 20 °C as determined from mass and volume measurements, the l 0 length of the sample under the same conditions, the Δl measured difference between the length l and T, and l 0. In most cases the third term on the right side of equation (1) is negligible. Figure 5 shows ΔV (%) versus temperature curves obtained from the relation:
As can be seen the thermal expansion seems to depend on the type and/or size of pendant side groups. Hydroxyl groups and acetyl groups differently influence on the network properties. Due to the presence of hydroxyl groups hydrogen bonds can be formed. Constituting an additional semi-bonded structure in a covalent-bonded network, they may increase its stiffness (crosslinking density). At some point, at higher temperatures when the crosslinks start to dislocate in the network, the hydrogen bonds would break gradually. Unbound hydroxyls, due to small size, should not interact as significantly with the neighboring chains between the crosslinks as acetyl groups larger in size. The latter apparently exert stronger effect on the network at higher temperatures. A possible mechanism of thermal volumetric expansion is depicted in Fig. 6. As opposed to the both-ends-connected network chains whose movements are restricted by the tangle of crosslinks, it is possible that the one-end-connected side groups and pendant unsaturations can partially change their position in the nearest space due to segmental rotations of atoms connected by single bonds. The rate of rotations would increase with the increasing temperature, and so would intensify the interactions with the neighboring chains. Being in a constant move the side groups would increase the range of their occupation in the surrounding space as the temperature increases. When enforcing the necessary room, they would cause the network to expand.

A possible mechamism of thermal volumetric expansion of a dimethacrylate network with pendant side groups (arrow size implies the extent of thermal motions)
Also, the presence or lack of an additional methyl group has its effect on the network. Having less rigid backbone the GMMA homopolymer, as opposed to the GDMA one, reaches its deflection point earlier. The presence of an additional methyl group in the Acet-GDMA increases the stiffness of the backbone homopolymer and as a consequence, the effect of “swelling” is less pronounced. So as to attain the same stiffness the acrylate/methacrylate homopolymer would have to reach a higher crosslinking density than its dimethacrylate counterpart. It may not be so, as proved by DMA and the double bond conversions which were found fairly equal for all homopolymers (see below). Interestingly, the hydroxyl-containing homopolymers, which have far more rigid networks than their acetylated analogs, do not show any anomaly in their HDT characteristics.
Conversion of unsaturated bonds
Conversion of double bonds in multifunctional methacrylates is rarely complete because of immobilisation, gelation, vitrification or steric isolation. In real systems, i.e., in chemically or lightly cured dimethacrylates, the conversion of double bonds ranges from 55 to 75 % [35, 36].
The di(meth)acrylate monomers used in this study can be considered as relatively low molecular weight crosslinking agents, in which methacrylate double bonds are separated by a short multiester chain. Usually, in order to find the conversion degree, intensities of peaks responsible for stretching vibrations of the C=C (1,637 cm−1) group before and after polymerization are compared. Depending upon the monomer type and comonomer composition, different peaks are used as internal standards.
In the present study, the polymerization conversion (DC) of the dimethacrylates was measured by DSC. The monomers were thermally cured with the use of BPO (1 wt%). To calculate the DC the following equation was used
where ΔH is the curing enthalpy of the sample, M w the molecular weight of a di(meth)acrylate, H m and H a the theoretical enthalpies of polymerization of one functional group—for the methacrylate double bond it equals 57 kJ/mol, for acrylate 82 kJ/mol [37], respectively, n m and n a—the number of methacrylic and acrylic groups per monomer molecule, and m—the weight of the sample.
The conversion degree dependence on the type of monomer is shown in Table 3.
So as to evaluate the DC, the heat of polymerization was measured in the range between 60 and 180 °C. As can be seen from the data in Table 3, all monomers are characterized by similar double bond conversions ranging 70–74 %. These values are presumed as maximum conversions attainable, and based on previous results [22] I hypothesize that the values of DC in networks cured by UV light and thermally post-cured should be fairly comparable. The acetylated monomers are characterized by slightly higher values of DC. This may be due to increased probability of cycle formation because of large pendant groups which additionally reduce the total amount of double bonds per sample weight/volume by increasing molecular weight. The values of DC for mono-acrylate monomers could be overestimated due to different reactivities of functional groups which were not accounted for in Eq. (3). These mean values were calculated assuming the “alternating” mode of polymerization of both polymerizable groups. Such assumption seems to be justified since more reactive radicals would preferably react with more reactive monomers. Nevertheless, one has to remember that the actual DC can differ from what was shown in Table 3.
Conclusions
In this study, it was shown how the presence of a pendant group may affect the thermo-mechanical properties of four different di(meth)acrylate networks. Dynamic mechanical measurements have demonstrated that the range of segmental and global mobilities in a crosslinked structure depends strongly on the nature of (meth)acrylate monomers used. Polymerization of monomers with the pendant hydroxyl groups as opposed to the acetyl groups seems to result in more heterogeneous networks. This heterogeneity might even deepen when more branched monomers are polymerized. Further, it was shown that the volume-temperature characteristics of a highly crosslinked structure depend strongly on the size/type of pendant groups. Large pendant groups and pendant unsaturations due to increased segmental mobility in respect to the immobile chains between crosslinks can cause a drastic increase in thermal expansion of the network. It is noteworthy that such network may increase its volume with temperature at faster rates than it increases its elasticity.
References
Anseth KS, Newman SM, Bowman CN. Polymeric dental composites: properties and reaction behavior of multimethacrylate dental restorations. Adv Polym Sci. 1995;122:176–217.
Kloosterboer JG. Network formation by chain crosslinking photopolymerization and its application in electronics. Adv Polym Sci. 1988;84:1–61.
Heath WH, Palmieri F, Adams JR, Long BK, Chute J, Holcombe TW, Zieren S, Truitt MJ, White JL, Wilson CG. Degradable cross-linkers and strippable imaging materials for step-and-flash imprint lithography. Macromolecules. 2008;41:719–26.
Cyr PW, Rider DA, Kulbaba K, Manners I. Photopatternable metallopolymers: photo-cross-linking and photolithography of polyferrocenylsilane methacrylates. Macromolecules. 2004;37:3959–61.
Nabais CRJOD, Heron BM, De Sousa HC, Gil MH, Sobral AJFN. Synthesis and characterization of co-polymers based on methyl methacrylate and 2-hexyl acrylate containing naphthopyrans for a light-sensitive contact lens. J Biomater Sci Polym Ed. 2011;22:139–52.
Barszczewska-Rybarek IM. Structure–property relationships in dimethacrylate networks based on Bis-GMA, UDMA and TEGDMA. Dent Mater. 2009;25:1082–9.
Kannurpatti AR, Anseth JW, Bowman CN. A study of the evolution of mechanical properties and structural heterogeneity of polymer networks formed by photopolymerizations of multifunctional (meth)acrylates. Polymer. 1998;39:2507–13.
Allen PEM, Williams DRG, Clayton AB. Dynamic-mechanical properties and cross-polarized, proton-enhanced, magic-angle spinning 13C-NMR time constants of urethane acrylates-2. Copolymer networks. Eur Polym J. 1994;30:427–32.
Simon GP, Allen PEM, Williams DRG, Cook WD. Properties of dimethacrylate copolymers of varying crosslink density. Polymer. 1991;32:2577–87.
Podgórski M, Matynia T. Network structure/mechanical property relationship in multimethacrylates—derivatives of nadic anhydride. J Appl Polym Sci. 2008;108:2902–13.
Matsumoto A, Ueda A, Aota H, Ikeda J. Effect of hydrogen bonds on intermolecular crosslinking reaction by introduction of carboxyl groups in free-radical crosslinking monomethacrylate/dimethacrylate copolymerizations. Eur Polym J. 2002;38:1777–82.
Matsumoto A. Free-radical crosslinking polymerization and copolymerization of multivinyl compounds. Adv Polym Sci. 1995;123:41–80.
Kloosterboer JG, Lijten GFCM. Thermal and mechanical analysis of a photopolymerization process. Polymer. 1987;28:1149–55.
Anseth KS, Bowman CN, Peppas NA. Polymerization kinetics and volume relaxation behavior of photopolymerized multifunctional monomers producing highly crosslinked networks. J Polym Sci Part A. 1994;32:139–47.
Cook WD. Photopolymerization kinetics of oligo (ethylene oxide) and oligo (methylene) oxide dimethacrylates. J Polym Sci Part A. 1993;31:1053–67.
Bowman CN, Peppas NA. Coupling of kinetics and volume relaxation during polymerizations of multiacrylates and multimethacrylates. Macromolecules. 1991;24:1914–20.
Yu Q, Zhou M, Ding Y, Jiang B, Zhu S. Development of networks in atom transfer radical polymerization of dimethacrylates. Polymer. 2007;48:7058–64.
Lienafa L, Monge S, Robin JJ. A versatile synthesis of poly(lauryl acrylate) using N-(n-octyl)-2-pyridylmethanimine in copper mediated living radical polymerization. Eur Polym J. 2009;45:1845–50.
Ide N, Fukuda T. Nitroxide-controlled free-radical copolymerization of vinyl and divinyl monomers. 2. Gelation. Macromolecules. 1999;32:95–9.
Cramer NB, Couch CL, Schreck KM, Boulden JE, Wydra R, Stansbury JW, Bowman CN. Properties of methacrylate-thiol-ene formulations as detal restorative materials. Dent Mater. 2010;26:799–806.
Avci D, Mathias LJ. Synthesis and photopolymerizations of new hydroxyl-containing dimethacrylate crosslinkers. Polymer. 2004;45:1763–9.
Podgórski M. Synthesis and characterization of novel dimethacrylates of different chain lengths as possible dental resins. Dent Mater. 2010;26:e188–94.
Lemon MT, Jones MS, Stansbury JW. Hydrogen bonding interactions in methacrylate monomers and polymers. J Biomed Mater Res A. 2007;83:734–46.
Andrzejewska E, Andrzejewski A. Sulfur-containing polyacrylates. IV. The effect of –S– and –O– linkages on the photo- and thermally induced polymerization of dimethacrylates. J Polym Sci Part A. 1993;31:2365–71.
Moszner N, Salz U. New developments of polymeric dental composites. Prog Polym Sci. 2001;26:535–76.
Podkościelna B. Photo- and thermally initiated polymerization of methacrylate monomer derivative of bis(4-hydroxyphenyl)sulfide with N-vinyl-2-pyrrolidone. J Therm Anal Calorim. 2010;. doi:10.1007/s10973-011-1628-0.
Podkościelna B. The highly crosslinked dimethacrylic/divinylbenzene copolymers. Characterization and thermal studies. J Therm Anal Calorim. 2011;104:725–30.
Podkościelna B, Worzakowska M. Synthesis, characterization, and thermal properties of diacrylic/divinylbenzene copolymers. J Therm Anal Calorim. 2010;101:235–41.
Podgórski M, Księżpolski J. Study of the effect of polymer network structure on thermal and mechanical properties of crosslinked (meth)acrylates–cis-hexahydrophthalic anhydride derivatives. Polimery. 2010;55:33–40.
Podgórski M. Synthesis and characterization of acetyloxypropylene dimethacrylate as a new dental monomer. Dent Mater. 2011;27:748–54.
Scott TF, Cook WD, Forsythe JS. Photo-DSC cure kinetics of vinyl ester resins I. Influence of temperature. Polymer. 2002;43:5839–45.
Jones DS. Dynamic mechanical analysis of polymeric systems of pharmaceutical and biomedical significance. Int J Pharm. 1999;179:167–78.
Podgórski M. Structure-property relationship in new photo-cured dimethacrylate-based dental resins. Dent Mater. 2012;28:398–409.
Heydemann P, Guicking HD. Specific volume of polymers as a function of temperature and pressure. Colloid Polym Sci. 1963;193:16–25.
Watts DC. Dental restorative materials. In: Williams DF, editor. Medical and dental materials. New York: VCH; 1992. p. 209.
Andrzejewska E. Photopolymerization kinetics of multifunctional monomers. Prog Polym Sci. 2001;26:605–65.
Pączkowski J. Photochemistry of Polymers. Theory and application, Chap 4. In: Andrzejewska E, editor. Photopolymerization. Toruń: Wydawnictwo Uniwersytetu Mikołaja Kopernika; 2003. p. 117.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Podgórski, M. Thermo-mechanical behavior and specific volume of highly crosslinked networks based on glycerol dimethacrylate and its derivatives. J Therm Anal Calorim 111, 1235–1242 (2013). https://doi.org/10.1007/s10973-012-2508-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2508-y