
Abstract
Hydrotalcite-like materials containing apart from magnesium and aluminum also copper, cobalt, nickel, and iron were prepared by a co-precipitation method. Thermal transformations of hydrotalcite-like materials were studied by thermal analysis methods as well as XRD, UV–vis–DRS, and XPS measurements of the samples calcined at various temperatures (600, 700, and 800 °C). Calcined hydrotalcites, especially those containing cobalt and copper, were found to be active and selective catalysts of N2O decomposition. It was shown that an increase in the calcination temperature significantly activated the Co-containing catalysts. Promotion of the samples with potassium resulted in activation of the hydrotalcite-based catalysts.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrotalcites are layered double hydroxides (LDHs) which show the brucite-like network characteristic of [Mg(OH)2]. In brucite, Mg2+ ions are octahedrally coordinated by hydroxyl anions, giving rise to the edge-shared layers. Part of the Mg2+ cations is replaced by trivalent ions (e.g., Al3+), resulting in the positive charging of the brucite layers. This charge is compensated by anions (e.g., CO3 2−) which together with water molecules are located in the interlayer space of LDHs. Furthermore, some of Mg2+, as well as Al3+ ions can be replaced, respectively, by other di- (e.g., Cu, Co, and Ni) and/or trivalent (e.g., Ni, Co, and Fe) cations with similar radius. The hydrotalcite-like structures can be obtained for a relatively large group of di- and trivalent metal cations in a broad range of these metal loadings [1]. Thermal decomposition of hydrotalcite-like materials at middle temperatures (T < 600 °C) results in the formation of the relatively high surface mixed metal oxides, with a high dispersion of introduced transition metals oxide species [2–6]. An increase in the calcination temperature results in the aggregation of these species and formation of the spinel phases [2–6]. Therefore, hydrotalcite-like materials are excellent precursors for the preparation of the metal oxide catalysts of the desired chemical and phase composition. A great number of scientific papers presenting the studies of hydrotalcite-based materials as catalysts for various chemical processes was published. For example, Cu–Mg–Al mixed oxides were found to be active and selective catalysts for the DeNOx process [3] and selective catalytic oxidation of ammonia to nitrogen and water vapor [7]. Hydrotalcite-based mixed metal oxides modified with nickel were effective catalysts of steam methane reforming [8], while iron containing hydrotalcites were recognized as precursors of effective catalysts for ethylbenzene dehydrogenation [9].
The article presents the studies of hydrotalcite-like materials containing apart from magnesium and aluminum various transition metals. The hydrotalcite precursors were calcined at various temperatures and therefore the metal oxides samples with the different phase composition were produced. The obtained samples were tested in the role of the catalysts for the N2O decomposition, which is very sensitive for the phase composition of the metal oxide systems.
The catalytic activity of various types of the metal oxides systems in the N2O decomposition was reported in the scientific literature. Cherlam et al. [10] showed high catalytic activity of Co3O4 spinel, which was additionally increased by partial substitution of another metal cation into the cobalt spinel structure (M x Co1−x Co2O4, M = Ni or Mg) [11]. Recently, Russo et al. [12], who tested various spinel systems, reported high catalytic activity of MgCo2O4 and ZnCo2O4 spinels, which were more catalytically active than Co3O4. The activation effect of the CeO2 addition in Co3O4 spinel was shown by Xue et al. [13]. The best catalytic results were obtained for the molar ratio of Ce/Co around 0.05. On the other hand, also copper was found to be an active component of the catalysts for N2O composition. Dandekar et al. [14] studied an influence of the kind of the support on catalytic activity of the Cu-containing catalysts. Centi et al. [15] reported the high activity of the copper–zirconia catalysts in N2O decomposition. The activity of these catalysts was comparable with that measured for ZSM-5 doped with copper. Also copper deposited on mesoporous silica was found to be the active catalyst for the N2O decomposition and reduction [16]. Hydrotalcite-like materials were reported to be very interesting precursors for synthesis of catalysts for N2O decomposition. Armor et al. [17] studied hydrotalcite-like materials containing apart from magnesium and aluminum also cobalt, nickel, rhodium, or palladium as precursors of catalysts for N2O decomposition. The best results were obtained for the Co–Al sample with the Co/Al ratio of 3. Calcined Co–Mn–Al hydrotalcites were studied by Obalova et al. [18–20], who proposed the optimal method for synthesis of effective catalysts and reported high activity of these oxide systems in the presence of O2 and NO x . Chang et al. [21] reported high activity of calcined Co–Al hydrotalcites containing additionally rhodium, palladium, or cerium in the process of N2O decomposition and reduction by carbon monoxide. Kannan and Swamy [22] reported that calcined Co–Al hydrotalcites obtained by supersaturation method are more active in N2O decomposition than the samples produced by sequential saturation method.
The analysis of the literature data shows that the process of N2O decomposition is very sensitive to kind and loading of catalytically active transition metal, its form, dispersion and used support.
Experimental
Catalysts preparation
Cu(II)Mg(II)Al(III), Co(II)Mg(II)Al(III), Cu(II)Co(II)Mg(II)Al(III), Ni(II)Mg(II)Al(III), and Mg(II)Fe(III)Al(III) hydrotalcite-like materials were prepared by the co-precipitation method using aqueous solutions of the following metal nitrates: Mg(NO3)2·6H2O (Sigma), Al(NO3)3·9H2O (Fluka), Cu(NO3)2·3H2O (Merck), Co(NO3)2·6H2O (POCh), Ni(NO3)2·6H2O (Acros), and Fe(NO3)3·9H2O (POCh). Solution of NaOH (POCh) was used as a precipitating agent. The mixture of metal nitrate solutions was slowly added to a vigorously stirred aqueous solution containing a slight excess of Na2CO3 (POCh). The pH was maintained constant at 10.0 ± 0.2 by dropwise addition of NaOH solution. An exception was Fe-containing hydrotalcite, which was synthesized at pH 8.5 ± 0.2. Precipitates were aged in a suspension at 60 °C for 30 min (or 40 °C for Fe-hydrotalcite) under vigorous stirring. In the next step, the suspension was filtered, washed with distilled water, and dried overnight at 120 °C. Finally, the prepared hydrotalcite-like materials were calcined at 600, 700, or 800 °C for 6 h. The samples codes and the molar ratios of metals are shown in Table 1. A detailed description of the hydrotalcite preparation was presented in our previous papers [2, 3].
The hydrotalcites calcined at 800 °C were modified with potassium using the incipient wetness impregnation method. Aqueous solutions of KNO3 (POCh) were used for the deposition of potassium. The intended potassium content in the samples was 0.09 and 0.9 wt%. The impregnated samples were calcined at 600 °C.
Catalysts characterization
For the selected hydrotalcite-like samples, the TGA–DTA measurements were performed using a Mettler Tolledo 851e thermobalance system. The experiments were performed in a flow of pure argon (80 mL/min) in the temperature range of 25–1,000 °C at a heating rate of 10 °C/min.
The surface area of hydrotalcite-like materials calcined at different temperatures was determined by the BET method. The measurements were performed using ASAP 2010 (Micromeritics). Prior to the nitrogen adsorption at −196 °C the samples were outgassed under vacuum at 350 °C for 12 h.
The X-ray diffraction (XRD) patterns of hydrotalcite-like materials and mixed metal oxides formed by their thermal decomposition were obtained with a Philips X’Pert APD diffractometer using Cu Kα radiation (λ = 1.54178 Å).
The coordination and aggregation of transition metals present in mixed metal oxides as well as their hydrotalcite precursors were studied by UV–vis–DR spectroscopy using an Evolution 600 (Thermo) spectrophotometer. The measurements were performed in the range of 200–900 nm with a resolution of 2 nm.
The surface composition of the calcined samples was studied by X-ray photoelectron spectroscopy (XPS). The measurements were performed at a room temperature in the ultrahigh vacuum (UHV) surface analysis system (Prevac). An Al Kα monochromatized radiation at 1486.6 eV and a hemispherical photoelectron energy analyzer (Scienta R3000) were used to determine the photoemission spectra. The XPS spectra were recorded with a pass energy of 100 eV. The data processing was performed with a CasaXPS, using a Shirley type background substraction and Gaussian/Lorentzian peak shapes. The binding energies were corrected by setting the Mg 2p peak at 50.5 eV, and the integrated intensities were corrected by atomic sensitivity factors.
Catalytic tests
The catalytic measurements of N2O decomposition were performed in a fixed-bed quartz microreactor at ambient pressure and in the range of temperatures 100–650 °C. The composition of outlet gas was analyzed on-line using quadrupole mass spectrometer RGA 200 (Prevac) connected directly to the reactor outlet. For each experiment, 0.1 g of a catalyst was placed on quartz wool in the reactor (6 mm i.d., l = 365 mm) and outgassed in a flow of pure helium at 500 °C. After cooling the reactor to 100 °C, the gas mixture containing 0.5 vol.% N2O and 4.5 vol.% O2 in He was fed into the reactor using mass flow controllers (Brooks). The total rate of gas flow was set at 50 mL/min and temperature was increased at a rate of 10 °C/min.
Results and discussion
Table 1 shows composition and BET surface area of hydrotalcites calcined at different temperatures. In a series of the samples calcined at 600 °C the highest surface area was measured for HT600-Cu15 and HT600-Co15. In general, an increase in the transition metal content increased surface area of the calcined samples (with exception of the samples containing both copper and cobalt), whereas and increase in calcination temperature decreased their surface area.
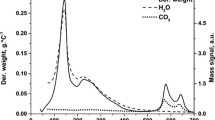
The thermal decomposition of selected hydrotalcites (HT-Cu15, HT-Co15, and HT-Cu10Co10) was studied using thermogravimetric method. The results of these studies are presented in Fig. 1. The thermal decomposition of hydrotalcites consists of few stages. A decrease of the samples weight that occurred at temperature below 250 °C, assisted by endothermic peak (DTA), is associated with the removal of the interlayer and weakly adsorbed water molecules [2]. For all the studied samples nearly this same weight loss (12.6–13.6 wt%) in this stage was detected. It could be seen that removal of water from the HT-Cu15 sample was easier than for HT-Co15. Therefore, it seems that water molecules are more loosely bound to brucite-like sheets containing Cu cations. The next stage of the thermal decomposition of hydrotalcites is observed in the temperature range of 250–500 °C. The loss of weight (~25%) is attributed to the decomposition of hydroxyl groups in the brucite-like layers, as well as the interlayer anions decomposition and removal. It could be seen that this stage of the hydrotalcite decomposition depended on the composition of the brucite-like sheets. In the case of the HT-Co15 sample, this process occurred in a relatively narrow temperature range of 220–420 °C. For the copper-containing samples (HT-Cu15 and HT-Cu10Co10), this stage of the hydrotalcites decomposition took place in a much broader temperature range from 210 °C to about 460–510 °C and proceeds in few steps. Two DTG peaks appeared for the Cu-containing samples at higher temperatures. As it was shown in our previous studies [2], the peaks at 660 and 565 °C found in the DTG curve of HT-Cu15 and HT-Cu10Co10 are related to the thermal decomposition of strongly stabilized structural CO3 2− anions. Similar effect was reported for Cu–Mg–Al hydrotalcites by Auer et al. [23]. It seems that copper introduced into the hydrotalcite structure stabilizes the CO3 2− anions, which are decomposed at higher temperature. The DTG peaks at about 800–850 °C are probably related to the thermal reduction of Cu(II) cations to Cu(I). Such effect was earlier reported for Cu–Mg–Al hydrotalcite derived mixed metal oxide even in oxygen containing atmosphere [3].

Results of thermogravimetric analysis of hydrotalcite-like materials
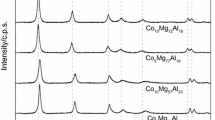
The XRD patterns of the as-prepared and calcined hydrotalcite-like materials are shown in Fig. 2. The diffractograms of the as-prepared samples (HT-Cu15, HT-Co15, and HT-Cu10Co10) are characterized by the presence of reflections typical of the hydrotalcite structure [2]. The comparison of the intensities of XRD reflections shows that crystallinity of the HT-Cu15 sample is higher than HT-Co15 and HT-Cu10Co10 ones. Calcination of the samples at 600 °C resulted in a disappearance of diffraction lines typical of the hydrotalcite structure and formation of three new broad reflexes. Two of them, positioned at about 42° and 62°, are characteristic of the MgO phase, whereas the band at about 37° is attributed to the presence of the spinel phase MgAl2O4 [2, 24]. For the cobalt containing samples (HT600-Co15 and HT600-Cu10Co10), the reflection at about 37° can be also related to the presence of the Co3O4 and CoO phases (Fig. 2a, c), whereas for the copper-containing samples (HT600-Cu15 and HT600-Cu10Co10) that peak could be also attributed the CuAl2O4 and CuO phases (Fig. 2b, c). An increase of the calcination temperature to 700 °C resulted in an appearance of new reflections characteristic of the mentioned above metal oxide and spinel phases, whereas in diffractograms of the samples calcined at 800 °C the sharp and intensive peaks typical of the metal oxide and spinel phases suggest a significant increase of their crystallinity.

Diffractograms of hydrotalcite-like samples calcined at different temperatures: h hydrotalcite, o metal oxides, s spinels
Therefore, it could be concluded that calcination of hydrotalcite-like materials at 600 °C results in their transformation into poorly crystalline mixed metal oxides. An increase in the calcination temperature to 700 °C results in the formation of the spinel phases, which become more ordered after calcination of the samples at 800 °C.
The UV–vis–DRS measurements were performed for hydrotalcite-like materials calcined at different temperatures. The samples were analyzed in the wavelength range of 200–900 nm at room temperature (Fig. 3). The as-synthesized copper-containing samples were blue, while hydrotalcites containing cobalt were pink. An increase in the cobalt content in the calcined samples resulted in a very deep green color (nearly black). To carry out the UV–vis–DRS measurements, the samples were diluted in commercial silica gel (silica gel/sample ratio was 9:1).

UV–vis–DR spectra of hydrotalcite-like materials calcined at different temperatures
The UV–vis–DR spectrum of the as-synthesized Co-containing hydrotalcite shows the specific band at around 520 nm (Fig. 3a). This absorption band is related to the presence of Co2+ cations in the octahedral coordination located in the brucite-like layers [25, 26]. This fact explains also the pink color of the samples. The broad band in the range of 600–700 nm can be found in the spectrum of the calcined cobalt-containing samples. This band is attributed to the d–d transitions in octahedrally coordinated Co3+ cations (transition 1T1g (I) ← 1Ag(I)) [26]. It should be noted that this absorption band is absent in the spectrum of as-synthesized hydrotalcite, so the thermal treatment (darker, nearly black color of the sample) results in a partial oxidation of Co2+ to Co3+. Probably, the increased calcination temperature provoked the Co3O4 spinel formation. The absorption band at around 370 nm, which appeared only in the spectrum of the calcined samples, also proves the existence of the Co3+ cations in the octahedral coordination (transition 1T2g (I) ← 1Ag (I)) [26]. Usually, the band at around 230–250 nm results from the oxygen to metal charge transfer [27].
Figure 3b shows the UV–vis–DR spectra recorded for a series of the Cu-containing samples. The spectrum of as-synthesized hydrotalcite shows the intensive band at about 225 nm, which can be attributed to the metal to ligand charge transfer (transition O2− ← Cu2+). The broad shoulder around 800 nm is associated with the Cu2+ transition in the octahedral coordination, while the band located at 680 nm is related to the Cu2+ transition in the deformed octahedral coordination [28]. The UV–vis–DR spectra of the calcined Cu-containing samples show the band at about 260 nm, which is related to the presence of Cu2+ and Al3+ in the spinel like structure (transition 2Eg ← 2T2g) [28]. The band at 330 nm could be assigned to the charge transfer between Cu2+ and oxygen in oligomeric metal oxide species [Cu–O–Cu] n . The broad band at around 435 nm is related to the d–d transfer in bulky CuO.
Figure 3c shows the UV–vis–DR spectra recorded for a series of the samples containing both cobalt and copper. It should be noted that these spectra are characterized by the presence of the bands typical of the transitions occurred in cobalt and copper species, which were found in the samples containing only one of these transition metals (cf. Fig. 3a, b).
The UV–vis–DRS studies proved that calcination of the hydrotalcite samples resulted in the formation of the aggregated metal oxide and spinel phases. These results are fully consistent with the results of XRD analysis of the samples.
XPS spectra were recorded for the selected samples (HT-Cu15 and HT-Co15) after calcination at 600 and 800 °C as well as modification with potassium. The collected results are summarized in Table 2. The calcined Cu- and Co-containing hydrotalcites showed peaks at 935.1–935.4 eV (Cu 2p3/2) and 780.3–780.6 eV (Co 2p3/2), respectively, characteristic of Cu2+ cations in CuAl2O4 [29] and Co2+/Co3+ in Co3O4 and CoAl2O4 [30]. It can be found that the content of transition metals at the sample surface, especially in the case of copper-loaded samples, is significantly lower compared to that in bulk. Such phenomenon was previously observed by Bridier et al. [31] for calcined Cu–Al hydrotalcites and was explained by segregation of transition metal penetrating the interior of alumina. An interesting effect was observed for the O 1s core level peak, which can be clearly deconvoluted into two components assigned to O2− lattice oxygen (at lower binding energy) and to oxygen of hydroxyl group (at higher binding energy). The contribution of lattice oxygen form increased both for the HT-Cu15 and HT-Co15 sample after raising the calcination temperature from 600 to 800 °C. An introduction of potassium to the catalyst calcined at 800 °C resulted in an increase of amount of hydroxyl groups present on the sample surface. Higher binding energies of O 1s photoelectron line measured for the Cu-doped samples compared to that characteristic of Co-containing ones confirm additionally enrichment of the HT-Cu15 sample in aluminum.
The calcined hydrotalcite-like materials were examined as catalyst for the N2O decomposition. Nitrogen and oxygen were the only detected products of this process. Figure 4 shows the N2O conversion measured in the presence of the catalysts containing various transition metals and calcined at 600 °C. The catalytic activity of these samples can be ordered as: HT600-Cu10 > HT600-Co10 > HT600-Fe10 > HT600-Ni10. As the copper- and cobalt-containing samples were found to be significantly more active catalysts compared to the Fe- and Ni-loaded ones, the detailed studies were focused on the optimization of their chemical and phase composition.

Results of catalytic tests performed for the samples containing Cu, Co, Fe, and Ni calcined at 600 °C
Figure 5 shows the results of the catalytic studies for the samples containing copper (Fig. 5a), cobalt (Fig. 5b), or both copper and cobalt (Fig. 5c) calcined at 600, 700, or 800 °C. Increasing the calcination temperature of the copper-containing sample from 600 to 800 °C significantly decreased its catalytic activity. An opposite effect was observed for the cobalt and copper–cobalt-containing samples. For a series of the HT-Co15 catalysts, an increase in the calcination temperature from 600 to 700 °C only slightly increased activity whereas a very significant activation of the catalyst occurred after calcination at 800 °C. For the catalysts containing both copper and cobalt a distinct improvement in the catalytic activity was observed for the sample calcined at 700 °C. An increase of the calcination temperature from 700 to 800 °C also resulted in the activation of the catalyst, however, this effect was not so significant. In the series of the catalysts calcined at various temperatures the highest activity was obtained for the copper–cobalt-containing sample calcined at 800 °C.

Results of catalytic tests performed for the samples calcined at 600, 700, and 800 °C, a HT-Cu15, b HT-Co15, and c HT-Cu10Co10
As it was shown by XRD, UV–vis–DRS, and XPS analysis, the phase composition and textural parameters of the samples strongly depends on calcination temperature. An increase in the calcination temperature favors the formation of the spinel phases and additionally significantly decreases the surface area of the samples. For a series of the HT-Cu15 catalysts, a decrease of activity was observed for the samples calcined at increased temperatures. This effect could be explained by a decrease of the surface area of the samples and possibly also by lower activity of copper incorporated into the spinel structures. An increase in the calcination temperature decreased also the surface area of the cobalt containing samples (HT-Co15 series), however, despite this effect the samples calcined at 700 and 800 °C were significantly more catalytically active than that calcined at 600 °C. This suggests that cobalt cations incorporated into the spinels structures are much more catalytically active than these present in the other forms. The interesting results were obtained for the samples containing both copper and cobalt. Taking into account the results obtained for the samples containing only one transition metal (Cu or Co) it could be expected that an increase in the calcination temperature should result in transformation of copper into less catalytically active species whereas an opposite effect should be observed for cobalt. Therefore, the expected catalytic activity of the HT800-Cu10Co10 sample should be between the HT800-Cu15 and HT800-Co15 catalysts whereas our studies showed the following activity order: HT800-Cu10Co10 > HT800-Co15 > HT800-Cu15. It seems that there is the synergetic effect related to the simultaneous presence of copper and cobalt oxides.
It was reported that promotion of the various transition metal oxide-based catalysts with alkali metals salts resulted in their activation in the N2O decomposition reaction [32]. The influence of potassium addition on the catalytic activity was studied for the hydrotalcite samples calcined at 800 °C. The results of catalytic tests performed for the HT800-Cu15, HT800-Co15, and HT800-Cu10Co10 samples and their modifications with different loading of potassium (0.09 or 0.9 wt%) are shown in Fig. 6. It should be noted that introduction of potassium activated the catalysts in the N2O decomposition. The most significant increase in the catalytic activity was found for the copper-containing sample. In this case, the potassium loading strongly influenced the activity of the catalysts. For the HT800-Co15 sample, deposition of lower amount of potassium (0.09 wt%) resulted in a slight deactivation of the catalyst, whereas a significant increase in activity was observed for the sample with higher content of potassium (0.9 wt%). An introduction of potassium into the HT800-Cu10Co10 catalyst improved its activity, however, the activation effect was less significant than in the case of the other samples. As the catalysts are characterized by various specific surface area the reaction rate measured at 450 °C and related to this parameter of the samples is presented in Table 3. It should be noted that the reaction rate increased after deposition of potassium, however, this effect was less significant for the samples promoted with 0.09% of potassium and much more significant for the catalyst modified with 0.9% of this alkali promoter. Promotion effect of potassium on decomposition of N2O over various metal oxide catalysts was reported in literature. Xue et al. [13] explained the increased activity of the potassium-doped Co3O4–CeO2 catalyst by improvement of the redox properties of Co3O4 by small amount of alkali metal. Similar explanation of activation effect was proposed by Asano et al. [32] for potassium-doped Co3O4 catalyst. However, in this case authors suggest that potassium promotes mainly reoxidation of Co2+ to Co3+. According to Obalova et al. [33], the promotion of the catalytic activity of the samples is due to an increase in the electron density on the Co sites by addition of potassium. This effect forces the electron donation to the adsorbed N2O, and avail fast N2O chemisorption and fast desorption of oxygen. Haneda et al. [34] reported that doping alkali metals into the cobalt oxide catalysts can weaken the Co–O bond strength and promote oxygen desorption from Co3O4. These authors suggested also that the low electronegativity of potassium may promote the electron transition from the active site to antibonding orbital of N2O and therefore facilitate the breaking the N–O bound. Similar mechanism of the potassium promotion effect was suggested by Cheng et al. [35] for the Co–Al catalysts prepared from hydrotalcite-like precursors.

Results of catalytic tests performed for the samples calcined in 800 °C, modified by different amount of potassium, a HTCu15, b HTCo15, and c HTCu10Co10
Conclusions
Magnesium–aluminum hydrotalcite-like materials containing additionally cobalt, copper, nickel, and iron were synthesized and calcined at various temperatures. It was shown that an increase in calcination temperature promoted the formation of the spinel phases. The calcined hydrotalcites were studied in the role of the catalysts for N2O decomposition. Among the samples calcined at 600 °C the best results were obtained for the copper-containing catalysts. An increase in the calcination temperature to 700 °C significantly activated the cobalt-containing catalysts, while the opposite effect was found for the samples containing copper. Among the samples calcined at 800 °C the best results were obtained for the catalysts containing simultaneously both cobalt and copper. The activation of the cobalt-containing samples was related to the formation of the catalytically active spinel phases. An additional activation effect was observed after promotion of the samples with potassium, however, the copper-containing samples were activated much more effectively than the other catalysts.
References
Cavani F, Trifirò F, Vaccari A. Hydrotalcite-type anionic clays: preparation, properties and applications. Catal Today. 1991;11:173–301.
Chmielarz L, Kuśtrowski P, Rafalska-Łasocha A, Dziembaj R. Influence of Cu, Co and Ni cations incorporated in brucite-type layers on thermal behaviour of hydrotalcites and reducibility of the derived mixed oxide systems. Thermochim Acta. 2003;395:225–36.
Chmielarz L, Kuśtrowski P, Rafalska-Łasocha A, Majda D, Dziembaj R. Catalytic activity of Co–Mg–Al, Cu–Mg–Al and Cu–Co–Mg–Al mixed oxides derived from hydrotalcites in SCR of NO with ammonia. Appl Catal B. 2002;35:195–210.
Ye L, Firdaus A. High temperature adsorption of carbon dioxide on Cu–Al hydrotalcite-derived mixed oxides: kinetics and equilibria by thermogravimetry. J Therm Anal Calorim. 2009;97:885–9.
Voyer N, Soisnard A, Palmer SJ, Martens WM, Frost RL. Thermal decomposition of the layered double hydroxides of formula Cu6Al2(OH)16CO3 and Zn6Al2(OH)16CO3. J Therm Anal Calorim. 2009;96:481–5.
Bakon KH, Palmer SJ, Frost RL. Thermal analysis of synthetic reevesite and cobalt substituted reevesite (Ni, Co)6Fe2(OH)16(CO3)·4H2O. J Therm Anal Calorim. 2010;100:125–31.
Chmielarz L, Kuśtrowski P, Rafalska-Łasocha A, Dziembaj R. Selective oxidation of ammonia to nitrogen on transition metal containing mixed metal oxides. Appl Catal B. 2005;58:235–44.
Christensen KO, Chen D, Lødeng R, Holmen A. Effect of supports and Ni crystal size on carbon formation and sintering during steam methane reforming. Appl Catal A. 2006;314:9–22.
Balasamy RJ, Khurshid A, Al-Ali AAS, Atanda LA, Sagata K, Asamoto M, Yahiro H, Nomura K, Sano T, Takehira K, Al-Khattaf SS. Ethylbenzene dehydrogenation over binary FeO x –MeO y /Mg(Al)O catalysts derived from hydrotalcites. Appl Catal A. 2010;390:225–34.
Cherlam U, Xu ZP, Zeng HC. Catalytic decomposition of N2O over M x Co1−x Co2O4 (M = Ni, Mg) spinel oxides. Chem Mater. 2000;12:650–8.
Yan L, Ren T, Wang XL, Ji D, Suo JS. Catalytic decomposition of N2O over M x Co1−x Co2O4 (M = Ni, Mg) spinel oxides. Appl Catal B. 2003;45:85–90.
Russo N, Fino D, Saracco S. N2O catalytic decomposition over various spinel-type oxides. Catal Today. 2007;119:228–32.
Xue L, Zhang C, He H, Teraoka Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl Catal B. 2007;75:167–74.
Dandekar A, Vannice MA. Determination of the dispersion and surface oxidation states of supported Cu catalysts. J Catal. 1998;179:111–28.
Centi G, Cerrato G, D’Angelo S, Finardi U, Giamello K, Morterra C, Perathoner S. Catalytic behavior and nature of active sites in copper-on-zirconia catalysts for the decomposition of N2O. Catal Today. 1996;27:265–70.
Chmielarz L, Kuśtrowski P, Kruszec M, Dziembaj R, Cool P, Vansant EF. Nitrous oxide reduction with ammonia and methane over mesoporous silica materials modified with transition metal oxides. J Por Mater. 2005;12:183–91.
Armor JN, Braymer TA, Farris TS, Li Y, Petrocelli FP, Weist EL, Kannan S, Swamy CS. Calcined hydrotalcites for catalytic decomposition of N2O in simulated process streams. Appl Catal B. 1996;7:397–406.
Pacultova K, Obalova L, Kovanda F, Jiratova K. Catalytic reduction of nitrous oxide with carbon monoxide over calcined Co–Mn–Al hydrotalcite. Catal Today. 2008;137:385–9.
Obalova L, Jiratova K, Kovanda F, Pacultova K, Lacny Z, Mikulova Z. Catalytic decomposition of nitrous oxide over catalyst prepared from Co/Mg–Mn/Al hydrotalcite-like compounds. Appl Catal B. 2005;60:289–97.
Obalova L, Pacultova K, Balabanova J, Jiratova K, Bastil Z, Valaskova M, Lacny Z, Kovanda F, Mikulova Z. Effect of Al/Mn ratio in Co–Mg–Al mixed oxide catalyst prepared from hydrotalcite-like precursors on catalytic decomposition of N2O. Catal Today. 2007;119:233–8.
Chang KS, Lee H-J, Park Y-S, Woo J-W. Enhanced performance of N2O destruction in the presence of CO over the mixed metal oxide catalysts derived from hydrotalcite-like precursors. Appl Catal A. 2006;309:129–38.
Kannan S, Swamy CS. Catalytic decomposition of nitrous oxide over calcined cobalt aluminium hydrotalites. Catal Today. 1999;53:725–37.
Auer SM, Wandeler R, Göbel U, Baiker A. Heterogeneous coupling of phenylethyne over Cu–Mg–Al mixed oxides: influence of catalyst composition and calcination temperature on structural and catalytic properties. J Catal. 1997;169:1–12.
Enache DI, Rebours B, Roy-Auberger M, Revel R. In situ XRD study of the influence of thermal treatment on the characteristics and the catalytic properties of cobalt-based Fischer–Tropsch catalysts. J Catal. 2002;205:346–53.
Perez-Ramırez J, Mul G, Kapteijn F, Moulijn JA. On the stability of the thermally decomposed Co–Al hydrotalcite against retrotopotactic transformation. Mater Res Bull. 2001;36:1767–75.
Herrero M, Benito P, Labajos FM, Rives V. Stabilization of Co2+ in layered double hydroxides (LDHs) by microwave-assisted ageing. J Solid State Chem. 2007;180:873–84.
Lim L, Ciuparu D, Park C, Dobek F, Chen Y, Harding D, Pfefferie L, Haller G. Synthesis and characterization of highly ordered Co-MCM-41 for production of aligned single walled carbon nanotubes (SWNT). J Phys Chem B. 2003;107:11048–56.
Crivello M, Perez C, Herrero E, Ghione G, Casuscelli S, Rodriguez-Castellon E. Characterization of Al–Cu and Al–Cu–Mg mixed oxides and their catalytic activity in dehydrogenation of 2-octanol. Catal Today. 2007;107–108:215–22.
Strohmeier BR, Leyden DE, Field RS, Hercules DM. Surface spectroscopic characterization of Cu Al2O3 catalysts. J Catal. 1985;94:514–30.
Chuang TJ, Brundle CR, Rice DW. Interpretation of the x-ray photoemission spectra of cobalt oxides and cobalt oxide surfaces. Surf Sci. 1976;59:413–29.
Bridier B, Lopez N, Perez-Ramirez J. Partial hydrogenation of propyne over copper-based catalysts and comparison with nickel-based analogues. J Catal. 2010;269:80–92.
Asano K, Ohnishi C, Iwamoto S, Shioya Y, Inoue M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl Catal B. 2008;78:242–9.
Obalova L, Karaskova K, Jiratova K, Kovanda F. Effect of potassium in calcined Co–Mn–Al layered double hydroxide on the catalytic decomposition of N2O. Appl Catal B. 2009;90:132–40.
Haneda M, Kintaichi Y, Bion N, Hamada H. Alkali metal-doped cobalt oxide catalysts for N2O decomposition. Appl Catal B. 2003;46:473–82.
Cheng H, Huang Y, Wang A, Li L, Wang X, Zgang T. N2O decomposition over K-promoted Co–Al. Catalysts prepared from hydrotalcite-like precursors. Appl Catal B. 2009;89:391–7.
Acknowledgements
This work was supported by the International PhD studies programme at the Faculty of Chemistry Jagiellonian University within the Foundation for Polish Science MPD Programme co-financed by the EU European Regional Development Fund. The research was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chmielarz, L., Rutkowska, M., Kuśtrowski, P. et al. An influence of thermal treatment conditions of hydrotalcite-like materials on their catalytic activity in the process of N2O decomposition. J Therm Anal Calorim 105, 161–170 (2011). https://doi.org/10.1007/s10973-011-1284-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-011-1284-4