
Abstract
A novel dilatometer has been used to measure the evolution of specific volume at different cooling rates and at elevated pressures under quiescent conditions and under shear for a series of commercial iPP homopolymers and polypropylene–ethylene random copolymers. Significant influences of cooling rates, pressures, and shear flow on the transition temperature T tr related to molecular weight and polydispersity and of course the temperature at which shearing was applied could be found for the iPP homopolymers. In the copolymers, the composition defined by the ethylene content determined the position of the transition temperature T tr.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Isotactic polypropylene can crystallize in three morphological forms; the α-, β-, and γ-phase differing in chain arrangements [1–4]. The monoclinic α-form is the most common crystalline phase, observed for both solution and melt crystallized samples at ambient pressure. The hexagonal β-modification of polypropylene is obtained sporadically at high supercoolings or the presence of selective β-nucleating agents [4, 5]. The triclinic γ-phase is favored by the presence of structural defects in the polypropylene chain that can be introduced either by incorporating very low molecular weight branches [6, 7] or by copolymerization with ethylene co-units of <10%mol [8–13]. Moreover, the crystallization of γ-phase is enhanced under high pressure [14–17]. Studies on crystallization of propylene–ethylene (P/E) copolymers showed that the amount of γ-phase increases with increasing ethylene content and crystallization temperature and with decreasing cooling rate [18], simultaneously resulting in a moderate decrease in crystallinity [19]. Ethylene units disturb the chain regularity, hindering crystal growth, and promoting chain folding. The special interest in studying this class of materials finds its origin in the improved transparency [20, 21], relative softness, and moderately improved low-temperature impact strength, characteristics that make these materials favorable for use in film, rigid packing, and pipe applications.
This article is about the PVT (pressure–volume–temperature) behavior of P/E random copolymers, focusing on the influence of ethylene content, cooling rate, and shear. The aim is to study the evolution of specific volume under (near-) processing conditions as found in, for example, injection molding. Morphological studies of the specimen give indication of the degree of different crystal states.
Materials
Three iPP homopolymers different in molecular weight and molecular weight distribution and three P/E random copolymers, with ethylene content of 3.4%mol, 5.2%mol, and 7.3%mol, respectively [22], were investigated. Isotactic polypropylene iPP1 had a molecular weight of M w ~ 310 kg mol−1 and a polydispersity index PDI of M w/M n ~ 3.4, iPP2 a molar mass of M w ~ 376 kg mol−1 and a PDI of M w/M n ~ 6.7 and iPP3 had M w ~ 466 kg mol−1 and M w/M n ~ 6.6. iPP1 and iPP3 were provided by Borealis (including their molecular characterization). iPP2 was a Basell product and used by the group of Prof. Titomanlio at Salerno University who determined the molecular data. All copolymer grades had same molecular characteristics: a molecular weight of M w ~ 310 kg mol−1, and a PDI of M w/M n ~ 3.4, measured by high temperature size exclusion chromatography (135 °C) using trichlorobenzene as a solvent. Material data were summarized in Table 1.
Experimental techniques
The experiments were performed with a custom made piston-die dilatometer (see Fig. 1) that allows not only for high cooling rates but also to apply high shear rates [23] using as test samples injection molded rings of m ~ 65 mg, which fit closely to the piston. Measurements were carried out in isobaric cooling mode using two experimental runs, one without (baseline run) and one with the sample inserted (sample run), resulting in relative specific volume measurements. The procedure used is shown schematically in Fig. 2: first heating up the sample to a temperature of 220 °C with an average heating rate of 10 °C min−1, next keeping that temperature for 10 min to delete previously formed structures, subsequently pressurize the sample to the level desired and, next, cool to a temperature of 40 °C. In the case of quiescent experiments, synthetic grease (Krytox GPL 207, Dupont, thermal-stable to a temperature of 290 °C) was used on the surface of the sample to avoid friction between sample and dilatometer wall. The grease did not react with the polymer as confirmed by DSC measurements of samples treated with grease and ones without grease. When shear was applied, no grease could be used, and in those cases the reading of volume after crystallizing in the solid state was somewhat less accurate. The cooling rate was determined by the flux, and cooling medium used was time dependent.

Schematic drawing of the pressure cell

Schematic drawing of the experimental procedure
DSC experiments were performed on all materials to determine melting temperature and degree of crystallinity using a TA Instrument Q1000 DSC. Samples of 3–5 mg, cut from pellets, were heated to a temperature of 220 °C with a heating rate of 10 °C min−1, held at this temperature for 5 min to erase the memory of the melt and subsequently cooled with a rate of 10 °C min−1. The heating run was repeated since a better heat transfer was guaranteed in the second experiment when the sample was more homogeneously distributed in the pan. Crystallinity was calculated from the enthalpy determined from the cooling curve using a value for the crystalline α-phase of 209 J g−1 [24].
Thin slices of 5 μm thickness were cut from dilatometer samples ex situ with a glass knife in a Leica RM 2165 Microtome and Cryo-unit Leica LN21 at a temperature of T = −30 °C using liquid nitrogen as cooling medium. From these slices, micrographs were taken with an Olympus BX 51 microscope combined with a Color View 3u digital camera.
Influence of cooling rate
Specific volume measurements were performed at cooling rates of 0.1 °C s−1, 1.5 °C s−1 and elevated pressures of 100 and 600 bar to study the evolution of crystallinity as indicated by the transition temperature T tr between melt and solid states. Lubricated sample rings were used in the experiments to avoid friction between sample and surfaces of the metal walls.
The evolution of crystallinity was measured by monitoring the specific volume, defined as:
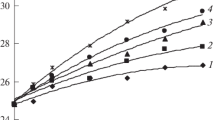
where Δx 1 was the axial die displacement measured in the experimental run, Δx 2 the axial die displacement recorded during the baseline run, r o the outer radius and r i the inner radius of the sample spacing, m the mass of the sample, C a constant, v 220_cf_Ref the absolute specific volume for the melt phase of the reference material iPP (K2XMOD, Borealis) [25] measured at a temperature of 220 °C with a CF-dilatometer and \( v_{{220\_{\text{PD}}}} \) the specific volume of the melt at a temperature of 220 °C measured with the PD-dilatometer. Since no reference data of measurements performed with a confined fluid dilatometer were available for a correct shift of all the materials used in this study, no absolute specific volume values were available. However, in this study, we focused on trends resulting from the molecular structure and thermo-dynamical history, and for this purpose, it proved sufficient to use reference data of one iPP grade (K2XMOD, Borealis) to shift our specific volume data to absolute values. This procedure seemed to be permitted since the slopes of the curves in the melt state region were the same for all materials investigated demonstrating that the isobaric thermal compressibility of a liquid or melt was only pressure and temperature dependent, which was already established by Zoller and Walsh [26]. Figure 3 shows results of quiescent experiments, used to calibrate the new dilatometer, applying cooling rates of 0.1 °C s−1 (top) and 1.5 °C s−1 (bottom) and enhanced pressures of P = 100 bar (left) and P = 600 bar (right) for iPP homopolymers (Fig. 3a) and for the P/E random copolymers (Fig. 3b). For the homopolymers, we found that the transition temperatures T tr shifted to lower temperatures with increasing cooling rate. iPP1 and iPP2 showed similar transition temperatures T tr while iPP3 crystallized at higher temperatures, independent of the pressure applied (see Table 2). From the morphology pictures in Fig. 4A, we learned that iPP1 and iPP2 had a comparable nucleation density (similar T tr and spherulite size, independent of the pressure applied), while iPP3 that crystallized at higher temperatures resulted in a more fine grained morphology. The nucleation density increased for increasing pressure for all three iPPs. No nucleation data were available at elevated pressures, but first experiments of the group of Eder and Janeschitz-Kriegl with these PP grades indicate that nucleation density indeed increases with increasing pressure [27]. For the copolymers, we found that the transition temperature T tr was directly related to the molecular structure of the polymer and shifted to lower temperatures with increasing ethylene content in the molecular chain. Furthermore, the transition temperatures were influenced by cooling rate in the same way as the homopolymers, i.e., shifting to lower temperatures with increasing cooling rate, confirmed by DSC experiments, see Table 1.

a Calibration experiment: Specific volume of iPP1, iPP2, and iPP3 samples at cooling rates of 0.1 °C s−1 (top) and 1.5 °C s−1 (bottom) at quiescent conditions, pressures 100 bar (left) and 600 bar (right). b As a, for the polypropylene–ethylene random copolymers

A Micrographs of samples arranged in the order iPP1, iPP2, and iPP3 (left to right). Cooling rates and pressures: a 0.1 °C s−1, 100 bar; b 0.1 °C s−1, 600 bar; c 1.5 °C s−1, 100 bar; d 1.5 °C s−1, 600 bar. B Micrographs of samples arranged in the order of increasing ethylene content from left to right starting with the homopolymer iPP1. Left side of each sample: piston wall; right side die wall. Cooling rates and pressures: a 0.1 °C s−1, 100 bar; b 0.1 °C s−1, 600 bar; c 1.5 °C s−1, 100 bar; d 1.5 °C s−1, 600 bar
The influence of pressure on specific volume was opposite, shifting the transition temperatures to higher values with increasing pressure, while changes in density were larger at low pressures. Higher end values of the specific volume were found for higher cooling rates caused by stresses introduced and frozen at higher cooling rates. The lower density implies a less perfect crystalline structure.
The resulting morphology was depicted in Fig. 4A and B, and generally showed sufficient homogeneity over the sample thickness, which indicated a homogeneous temperature distribution. At a cooling rate of 0.1 °C s−1, we found that the homopolymers and the 3.4%mol random copolymer RaCo1 developed spherulites with positive overall birefringence, RaCo2 (5.2%mol ethylene) appeared to be a borderline copolymer for the development of spherulitic structures forming circular spherulites as well as irregular objects, while the copolymer with the highest ethylene content of 7.3%mol, RaCo3, organized in irregular small bundles of crystallites, see Fig. 4B(a). The positive sign of the birefringence observed in all copolymers was related to their molecular structure and, consequently, their fractional content of γ-crystals [10]. A quantitative evaluation of the γ-phase content in the materials using X-ray measurements is part of a future investigation. At higher cooling rates, of 1.5 °C s−1, only the neat polymer forms spherulites with a high nucleation density, see Fig. 4B(c). RaCo1 was now the borderline copolymer, while RaCo2 and RaCo3 also showed irregular objects that appeared to be dendritic and lacked organized radial growth, which was an early indication of quenching into mesomorphic form [28].
Pressure-dependent structural changes were only evident for the homopolymer and RaCo1 at the low cooling rate of 0.1 °C s−1. The structure of the neat polypropylene at 100 bar [Fig. 4B(a)] developed large spherulites with low nucleation density and a negative birefringence, which changed to spherulitic morphologies of positive overall birefringence and higher nucleation density at pressures of 600 bar [Fig. 4B(b)]. RaCo1 formed, at elevated pressures of 100 bar, a spherulitic structure of circular spherulites and irregular objects, and a highly positive birefringent spherulitic structure with lower nucleation density at elevated pressures of 600 bar. For the other experimental conditions of the homopolymer and RaCo1, no morphological changes could be observed. RaCo2 and RaCo3, in general, showed no visible influence of pressure on structure development.
Influence of shear
Shear experiments were performed at the high cooling rate of 1.5 °C s−1 applying a shear rate step (with a shear rate of \( \dot{\gamma } = \, 67.5\;{\text{s}}^{ - 1} \, \) and a total shear of \( \gamma = 135 \)) at two different temperatures of T = 142 and 164 °C. In order to compare shear and quiescent experiments, a normalized specific volume v* was plotted, defined as
with v s the specific volume at ambient temperature and v m the specific volume in the melt state at 220 °C.
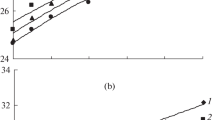
For comparison reason, the normalized specific volume of the quiescent experiments was plotted in Fig. 5 (bottom) while the shear experiments were summarized in Fig. 5 (top and middle) for the shear temperature of 164 and 142 °C, respectively. Some experiments showed some irregularities in the solid state or deviations in the melt state, caused by friction between the sample and surfaces of the metal walls since no lubricant could be used in shear experiments. Specific volume measurements were, however, rather sensitive in this dilatometer, since the total displacement range covered by an experiment was typically 0.5 mm and a deviation of 10 μm in displacement reading will already clearly show up. However, a deviation from the absolute value of the specific volume had no effect on the influence of shear on the transition temperature and, therefore, qualitative conclusions on the transition could still be drawn.


a Influence of shear. Relative specific volume of iPP1, iPP2, and iPP3 at a cooling rate of 1.5 °C s−1, a shear step (total shear \( \gamma = 135 \)and a shear rate \( \dot{\gamma } = \, 67.5\;{\text{s}}^{ - 1} \, \)) applied at temperature of T = 162 °C (top) and at T = 141 °C (middle), pressures of 100 bar (left) and 600 bar (right); (bottom) idem without shear. b As a, now for the polypropylene–ethylene random copolymers
Comparing quiescent measurements (Fig. 5, bottom) and shear experiments performed at T s = 164 °C and pressures of 100 and 600 bar (Fig. 5, top), it could be observed that the crystallization kinetics of the neat polypropylene were enhanced by flow, whereas the copolymers were hardly affected due to re-melting of the precursors. An exception was RaCo2 that showed some influence of flow on the transition temperature. A possible explanation for this could be the fact that this material was not pure and contained 0.2%wt of an antiblocking agent, which could slightly enhance the crystallization kinetics in flow experiments [29]. For these reasons, the results of the RaCo2 have to be treated with care when used in direct comparison to the other polymers.
At pressures of 100 bar, the transition temperature of the iPP1 grade increased from T tr = 124 °C at quiescent conditions to T tr = 133 °C in shear experiments with a shear step applied at a temperature of 142 °C and T tr = 134 °C for shear experiments at temperatures of 164 °C. It was obvious that the enhancement of the crystallization kinetics due to flow was limited in this case since the precursors formed in shear experiments at T s = 142 °C and even more at T s = 164 °C re-melt. For iPP2, the transition temperature increased from T tr = 122 °C to T tr = 138 °C to T tr = 152 °C for quiescent, shear experiments performed at T s = 142 °C and shear experiments at T s = 164 °C, respectively. Due to the higher polydispersity index of iPP2, i.e., a more pronounced high molecular weight tail at nearly the same average molecular weight, the effect of flow on crystallization kinetics increased compared to iPP1 and less precursors re-melted in the shear experiments indicated by a higher transition temperature. In the case of iPP3, the melt almost immediately started to crystallize after the application of flow, which proved that higher molecular weight and higher polydispersity were most effective in the enhancement of crystallization kinetics in flow experiments. Experiments performed at elevated pressures of 600 bar also showed an increase of transition temperature due to flow (Table 2). The melts in shear measurements at T s = 142 °C immediately started to crystallize after cessation of flow or even during flow (iPP3), but when the shear step was applied at T s = 164 °C only iPP2 and iPP3 immediately crystallized, while the precursors in iPP1 re-melted until a temperature of 151 °C. It should be remarked that the measurement of iPP3 in Fig. 5a showed an undershoot of the specific volume, which was physically not correct, but could be explained by the experimental setup. The pressure cell of the dilatometer was mounted on a big cone bearing, that showed some elasticity when a melt solidifies during shear experiments and the bearing gave temporarily way.
The transition temperatures measured were listed in Table 2 and hardly any differences were found between RaCo1 and RaCo2 when exposed to flow, despite the higher amount of chain defects in RaCo2. A quantitative separation of these two materials was found at more severe flow conditions, i.e., pressures higher than 400 bar and applying the shear at T s = 142 °C. At these conditions, the crystallization enhancing effect of the additive in RaCo2 could not compensate for the negative influence of the ethylene content on the crystallization kinetics.
Shearing at low temperatures was generally rather effective and the transition temperatures shifted to values about 10 °C higher than those in quiescent experiments (Fig. 5, bottom). Since, however, crystallization of all polymers studied still started at temperatures below the shearing temperature T s, part of the shear-induced precursors re-melted. Differences between polymers with different molecular structure became more obvious at higher pressures of 600 bar. The neat polypropylene grade immediately crystallizes after cessation of flow (crystallization time t cr = 2 s) and it is followed by RaCo1, which starts to crystallize at the same transition temperature but needed about 14 s to crystallize. Crystallization time (a direct measure for enhanced kinetics) was defined as the time difference between reaching the transition temperature and the solid state:
Transition temperatures were listed in Table 2, and the crystallization times were in Table 3. RaCo2 and RaCo3 both started at about the same transition temperature of T r = 135 °C and had crystallization times of t cr = 14 s.
Conclusions
A custom made dilatometer was used to investigate the influence of shear flow, cooling rate, and pressure on the evolution of specific volume of a series of iPP homopolymers and P/E random copolymers at non-isothermal conditions and elevated pressures. Shear flow, cooling rate, and pressure had a pronounced effect on specific volume, changing the transition temperature and the rate of transition. Higher cooling rates lead to a shift to lower temperatures and result in a lower density of the polymer. Increase of pressure led to higher nominal values of transition temperature. Morphology studies showed homogeneous temperature distribution in the samples and increasing nucleation density with increasing cooling rates. In general, the transition temperature shifted to higher temperatures in shear experiments, and the effect of flow was directly related with the increase of pressure, lower shear temperature, where the shear step was triggered, higher polydispersity and higher molecular weight. Shear steps applied at temperatures of 164 °C showed no effect of the copolymer composition, due to re-melting of shear-induced precursors. Flow at other temperatures showed a pronounced effect and the number of molecular defects (caused by the ethylene content) counteracted flow influences. Thus, copolymerization could be used to at least partly decouple flow effects and cooling rate effects.
References
Turner-Jones A, Aizlewood JM, Beckett DR. Crystalline forms of isotactic polypropylene. Makromol Chem. 1964;75:134–58.
Brückner S, Meille SV, Petraccone V, Pirozzi B. Polymorphism in isotactic polypropylene. Prog Polym Sci. 1991;16:361–404.
Addink EJ, Beintema J. Polymorphism of crystalline polypropylene. Polymer. 1961;2:185–93.
Varga J. Supermolecular structure of isotactic polypropylene. Mat Sci. 1992;27:2557–79.
Zhou G, He Z, Yu J, Han Z, Shi G. Studies on the β-form of isotactic polypropylene 1: characterization of the β-form and study of the β-α transition during heating by wide angle X-ray diffraction. Macromol Chem. 1986;187:633–42.
Lotz B, Graff S, Wittmann JC. Crystal morphology of the γ (Triclinic) phase of isotactic polypropylene and its relation to the α phase. J Polym Sci. 1986;24:2017–32.
Kojima MJ. Solution-grown lamellar crystals of thermally decomposed isotactic polypropylene. J Polym Sci B. 1967;5:245–50.
Turner-Jones A. Developments of the γ-crystal form in random copolymers of propylene and their analysis by DSC and X-ray methods. Polymer. 1971;12:487–507.
Alamo RG, Ghosal A, Chaterjee J, Thompson KL. Linear growth rates of random propylene ethylene copolymers. The changeover from γ dominated growth to mixed (α+γ) polymorphic growth. Polymer. 2005;46:8774–89.
Hosier IL, Alamo RG, Lin JS. Lamellar morphology of random metallocene propylene copolymers studied by atomic force microscopy. Polymer. 2004;45:3441–55.
Mezghani K, Phillips PJ. γ-Phase in propylene copolymers at atmospheric pressure. Polymer. 1995;35:2407–11.
Margio A, Causin V, Marega C, Ferrari P. Crystallization of the γ-form in random propylene-ethylene copolymers. Polym Int. 2004;53:2001–8.
Dimeska A, Phillips PJ. High pressure crystallization of random propylene-ethylene copolymers: α-γ phase diagram. Polymer. 2006;47:5445–56.
Campbell RA, Phillips PJ. The gamma phase of high-molecular-weight polypropylene. 1. Morphological aspects. Polymer. 1993;34:4809–16.
Mezghani K, Phillips PJ. The γ-phase of high molecular weight isotactic polypropylene II: the morphology of the γ-form crystallized at 200 MPa. Polymer. 1997;38:5725–33.
Kardos JL, Christiamsen AW, Baer E. Structure of pressure-crystallized polypropylene. J Polym Sci A. 1966;2(4):777–88.
Brückner S, Phillips PJ, Mezghani K, Meille SV. On the crystallization of γ-isotactic polypropylene: a high pressure study. Macromol Rapid Commun. 1997;18:1–7.
Laihonen S, Gedde UW, Werner PE, Martinez-Salazar J. Crystallization kinetics and morphology of poly(propylene-stat-ethylene) fractions. Polymer. 1997;38:361–9.
Laihonen S, Gedde UW, Werner PE, Westdahl M, Jääskelainen P, Martinez-Salazar J. Crystal structure and morphology of melt-crystallized poly(propylene-stat-ethylene) fractions. Polymer. 1997;38:371–7.
Resch K, Wallner GM, Teichert C, Maier G, Gahleitner M. Optical properties of highly transparent polypropylene cast films: influence of material structure, additives and processing conditions. Polym Eng Sci. 2006; 46:520–31.
Wallner GM, Resch K, Teichert C, Gahleitner M, Binder W. Effect of material structure and additives on the optical properties of PP cast films. Monatshefte für Chemie. 2006;137:887–97.
Gahleitner M, Jääskeläinen P, Ratajski E, Paulik C, Reussner J, Wolfschwenger J, et al. Propylene-ethylene random copolymers: comonomer effects on crystallinity and application properties. J Appl Polym Sci. 2005;95:1073–81.
Forstner R, Peters GWM, Meijer HEH. A novel dilatometer for volumetric dilatometry of polymers at high cooling- and shear rates. Int Polym Process. 2009;XXIV(2):114–21.
Brandrup J, Immergut EH, editors. Polymer handbook. 3rd ed. New York: Wiley; 1989.
van der Beek MHE, Peters GWM, Meijer HEH. A dilatometer to measure the influence of cooling rate and melt shearing on specific volume. Int Polym Process. 2005;XX(2):111–20.
Zoller P, Walsh DJ. Standard pressure-volume-temperature data for polymers. Lancaster, PA: Technomic Publishing Company Inc; 1995.
Eder G, Rexhepaj E, Ratajski E. Proceedings IUPAC World Polymer Congress MACRO2006, July 16–21, 2006, Rio de Janeiro, Brazil.
Foresta T, Piccarolo S, Goldbeck-Wood G. Competition between α and γ phases in isotactic polypropylene: effects of ethylene content and nucleating agents at different cooling rates. Polymer. 2001;42:1167–76.
Hwang WR, Peters GWM, Hulsen MA, Meijer HEH. Flow-induced crystallization of particle-filled-polymers. Macromolecules. 2006;39(24):8389–998.
Acknowledgements
The authors thank the Dutch Polymer Institute (DPI) and Moldflow for giving their financial support to this project and Prof. Guiseppe Titomanlio from Salerno University and Dr. Markus Gahleitner from Borealis Group for providing materials and molecular data.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Forstner, R., Peters, G.W.M., Rendina, C. et al. Volumetric rheology of polymers. J Therm Anal Calorim 98, 683–691 (2009). https://doi.org/10.1007/s10973-009-0552-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-009-0552-z