
Abstract
Both affordability and stability are important for commercial-scale production and industrial applications of TiO2. In addition, the ability to tailor nanostructure and physicochemical properties can provide advantages for future applications. Herein a facile sol‒gel process was investigated by using titanyl sulfate as an inexpensive feedstock reacting with water in the media of acetic acid and isopropanol. An anatase phase was readily produced at 65 °C, followed by drying at 80 °C. The anatase was stable up to 800 °C due to the residual sulfate and nitrogen, where sulfate and ammonium slowly decomposed when heating beyond 400 °C. The monolithic TiO2 xerogels were composed of agglomerated TiO2 spherical particles with diameters of ca. 50 or 100 nm. The TiO2 spherical particles were built by anatase crystallites with a diameter of ca. 5 nm. As a result, the TiO2 exhibited both bimodal mesopores and macropores: Large mesopores (10‒30 nm) were present due to the void spaces between the TiO2 spherical particles, while the smaller mesopores (ca. 3 nm) were due to the void spaces between the anatase crystallites within each TiO2 particle. There were also larger macropores (a few micrometers), which were caused by gas bubbles generated during the sol‒gel reactions. From a mass transfer viewpoint, these large pores within TiO2 xerogels could have advantages in their potential applications for catalysis and/or filtration processes.
Graphical Abstract

A thermal stable anatase TiO2 with large mesopores synthesized via sol‒gel reactions from TiOSO4.
Highlights
-
Sol‒gel synthesis of TiO2 using inexpensive industrial feedstock.
-
The anatase phase of TiO2 is thermal stable up to 800 °C.
-
The bimodal mesopores are potentially beneficial for mass transfer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
As an emerging semiconductor for solar energy harvest [1,2,3], the photocatalyst for hydrogen production [4], industrial catalysts [5, 6], and an environmental remedy [7], titania (TiO2) nanomaterials have been extensively studied. For example, TiO2 is catalytically more active than Al2O3 in the hydrolysis reaction of CS2 due to its oxygen deficiency (Eq. (1)) [6, 8]; however, the activity is also highly related to its porosity, surface area, and potential dopants. When capillary condensation of certain species occurs (such as water and elemental sulfur) in a certain process, there will be a higher resistance to mass transfer, and reactant/product diffusion will be limited to access/leave the active sites. In this circumstance, a larger pore size will be essential for maintaining the catalytic activity [9, 10]. In addition, large pore size is also important for the deposition of secondary and tertiary species, especially with a high loading, when TiO2 is used as catalyst support [11, 12].
Anatase and rutile are two dominant crystalline phases among TiO2 polymorphs, which also include brookite and TiO2(B) [13,14,15]. While anatase plays a significant role in its application as photocatalysts and photovoltaic devices, rutile is used more for pigment, sunlight-blocking products, and optical communication electronics [16].
Anatase is a more desirable phase (if kinetically stable), while rutile is the more thermodynamically stable crystalline phase, thus, the phase transition from anatase to rutile is irreversible. From a chemistry viewpoint, this solid-solid phase transition to rutile requires breaking some Ti‒O bonds, where literature reports a wide range of activation energies (20‒837 kJ mol−1, depending on particle size, impurity, etc.) [16]. The transition inevitably involves the growth of rutile crystallites and consequent collapse of micro- and mesopore structure, in addition to a decrease in surface area and an increase in the bandgap. Therefore, it is essential to know the kinetic crystalline stability when TiO2 is desired for a catalyst, catalyst support, and photocatalyst, especially at an elevated temperature and in the presence of steam [17].
The bench scale synthesis methods include sol‒gel [7], hydrothermal [18], self-assembly [19], templating [20], electrospinning [21], and chemical vapor deposition [22]. In these laboratory synthesis routes, titanium alkoxides or titanium chloride (TiCl4) are commonly used as starting materials. As an important industrial product for pigment and catalysis applications, TiO2 has been produced from the ilmenite and rutile minerals. The ore extraction strategies are limited to either the chloride or sulfate route, where a soluble intermediate of TiCl4 or TiOSO4, respectively, is formed [23, 24]. The oxidation processing of TiCl4 has been used to produce benchmark TiO2 nanoparticles, Degussa P25, which are extensively studied for their photocatalysis applications. There is an interest to use TiOSO4 to make TiO2 with desired nanostructures because TiOSO4 is significantly less expensive than titanium alkoxides [24].
Sol‒gel reactions of titanium alkoxides or salts have been extensively studied in the past decades, which include either aqueous or non-aqueous processes [25, 26]. The direct TiO2 products are often amorphous with some exceptions [27], but they can change into the anatase phase after calcination at 400 °C. The anatase phase normally starts to transform to rutile at 500 °C [28]. Therefore, a variety of research has been conducted to make anatase that is kinetically stable up to very high temperatures, e.g., 1000 °C [29,30,31].
In our previous research, anatase TiO2 was synthesized in different reaction media to control the morphology and pore structure for different purposes [32, 33]. However, the starting materials have been titanium alkoxides, which are expensive for commercial-scale production. The inexpensive precursor, TiOSO4 has been explored previously for making TiO2 [34,35,36,37,38,39], but very few studies have been reported on the crystalline stability and pore structure control of the final products [40, 41]. Herein, we explored a new sol‒gel procedure for making stable anatase with a large pore size by using TiOSO4 as the starting material, which is an inexpensive feedstock used for making non-sol-gel TiO2. The thermal stability of the resulting material was compared with the anatase prepared from titanium isopropoxide. The resulting materials have been of interest for both a Claus catalyst and catalyst support for hydrogenation desulfurization reactions.
2 Methods
2.1 Materials
97% Ti(OiPr)4, 28.0‒30.0% aqueous NH4OH, and 99.5% acetic acid (HAc) were purchased from Sigma−Aldrich, 99.5% isopropanol (IPA) was purchased from Fisher Chemicals, and 67.3% TiOSO4 (containing 7.2% H2SO4 and 25.5% water) was purchased from Alfa Aesar. These materials were used without further purification.
2.2 Synthesis of TiO2
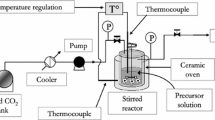
The sol‒gel synthesis was carried out using the experimental setup that has been described previously [42]. For the typical synthesis of TiO2 from TiOSO4, 72.17 g IPA, 72.12 g HAc, and 47.54 g TiOSO4 were added to a 2 L 3-neck flask while stirring. The slurry was heated to 65 °C and then 72 mL of deionized water was added. The solid was dissolved within 2 h and a clear solution was obtained (pH was measured as 1.6). Into this acidic solution, NH4OH was added dropwise till the pH reached the target level. Both gas and precipitate appeared upon and after the addition of NH4OH. The obtained material was aged for 2 days at 65 °C, followed by filtration and drying at 80°C under vacuum. Following this procedure, a series of TiO2 samples was synthesized as shown in Table 1. In the labels of TiO2 samples, the first letter “s” was for sulfate from the precursor TiOSO4, and the number represented the pH after the addition of NH4OH.
To study the solvent’s effect, the hydrolysis of TiOSO4 was conducted in isopropanol and water (without acetic acid) while other synthesis parameters were kept the same as for making TiO2-s-4.3, and the resulting material was called TiO2-i-4.3 (where “i” stands for isopropanol). Similarly, when acetic acid and water were used as the solvents (without isopropanol), the resulting material was called TiO2-a-4.3 (where “a” stands for acetic acid). In addition, TiO2 was also prepared without acetic acid and isopropanol, and the resulting material was called TiO2-w-4.3 (where “w” stands for water). The molar ratios for making each material are summarized in Table 1.
For comparison, TiO2 was also synthesized from Ti(OiPr)4 at room temperature (~22 °C). 200.00 g of Ti(OiPr)4 was dissolved in 694 mL IPA, and then 104 mL of 1 M HNO3 was added dropwise. At this point, the pH was measured at 2.2. The cloudy material was kept at room temperature for two days, followed by filtration and drying at 80 °C under a vacuum. The resulting material was labeled as TiO2-p-2.2-ap, where the first suffix “p” stood for isopropoxide from the precursor Ti(OiPr)4, and the second suffix represented the pH, and the third suffix “-ap” was for as -prepared sample. To identify the calcined samples, a suffix was added to indicate the calcination temperature, such as –400 for calcination at 400 °C. In a separate experiment, TiO2 was synthesized following the procedure for making TiO2-p-2.2, but NH4OH was used to adjust the pH from 2.2 to 7.0. This new sample was labeled as TiO2-p-7.0, as summarized in Table 2.
2.3 Characterization
N2 physisorption was conducted using a 3Flex (Micromeritics) instrument. The samples were activated in situ at 200 °C until the vacuum reached 1.0 × 10−4 torr. The reported pore size and pore size distribution were based on the desorption isotherm calculated using a BJH model. XRD was analyzed using a Rigaku Ultima IV diffractometer at a speed of 2° min−1 and a step size of 0.02°. XPS analyses were carried out with a Kratos AXIS Supra X-ray photoelectron spectrometer using a monochromatic Al K(alpha) source (15 mA, 15 kV). It probes the sample’s surface to a depth of 7–10 nanometers and has detection limits ranging from 0.1 to 0.5 atomic percent depending on the element. The instrument work function was calibrated to give binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold, and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The Kratos charge neutralizer system was used on all specimens. Survey scan analyses were carried out with an analysis area of 300 × 700 microns and a pass energy of 160 eV. High-resolution analyses were carried out with an area of 300 × 700 microns and a pass energy of 20 eV. Spectra have been corrected to the main line of the carbon 1 s spectrum (adventitious carbon) set to 284.8 eV. Spectra were analyzed using CasaXPS software (version 2.3.14). SEM images were collected on an FEI Philips XL30. The HRTEM images were obtained using a Tacnai 20 operated at 200 kV. The thermal destruction of sulfate in TiO2 was measured on a TA TGA 550 system. The samples were heated from 23 to 800 °C with a ramp of 5 °C min−1 under a flowing helium atmosphere (10 mL min−1). IR studies on solid samples were carried out on a Varian 7000 FTIR at a resolution of 4 cm−1. Ion chromatography was used to estimate the concentration of sulfate on the surface of TiO2 samples. The samples were prepared using 0.10 N NaOH to extract sulfate and sulfite from the spent catalysts using sonication. All the processes were conducted under an inert environment in an N2 glove bag to prevent the oxidation of sulfur compounds by air. GC analysis was conducted on a Varian CP-3800 equipped with two columns (a molsieve 5A and a Restek RT-U-Bond Plot) and two TCD detectors.
3 Results and discussion
3.1 Synthesis
TiOSO4 was found to be insoluble in the mixture of IPA and HAc (with a 1:1 molar ratio) in the temperature range of 22‒70 °C. Unlike titanium alkoxide, titanium (IV) in TiOSO4 was unable to react with HAc. The addition of water (from 72 to 144 mL) in the mixture at 65 °C could slowly dissolve TiOSO4 to form a clear solution, and then it became opaque within 2 h. The 2 h precipitation time at pH = 1.6 is significantly less than the precipitation time at pH = 1.4 and 1.0 (6 days and 45 days, respectively) [34]. This is because Ti salt hydrolysis depends on hydroxide concentration. Indeed, after the addition of NH4OH into the mixture of TiOSO4‒IPA‒HAc, precipitate and gas bubbles appeared immediately.
3.2 IR
All TiO2-s intermediates and products showed significant stretching modes for Ti‒O‒Ti oxo bonds at 570‒660 cm−1. There were also chemi- or physisorbed water bands at 3200‒3300 cm−1 and ~1630 cm−1, and residual sulfate at 1050‒1240 cm−1 (blue curve in Fig. 1) [43, 44]. To our surprise, the spectra did not show carboxylic groups at ca. 1720 cm−1. The absence of carboxylate ligands, but the presence of sulfate, suggested that the acetate ligand could not replace sulfate to form Ti-acetate complexes. The oxo bands could also be found with the TiO2-s made from TiOSO4 reacting with HAc in IPA only (i.e., without adding H2O and NH4OH), even though the oxo intensity is lower (black curve in Fig. 1). This result suggested that the sol‒gel process could occur under strongly acidic conditions and with a relatively small amount of water (from the starting material of TiOSO4, which contains 25.5% water). However, its morphology is more like the starting material TiOSO4 than the sol‒gel product TiO2 (see the SEM image in the SEM section).

IR spectra of TiOSO4 (brown), the product of TiOSO4 reacting with HAc in IPA (w/o addition of H2O and NH4OH) at 65 °C for 2 days (black), the as-prepared Ti-s-2.7 (light blue), the as-prepared TiO2-a-4.3 (dark blue), the as-prepared TiO2-i-4.3 (light brown), the as-prepared TiO2-w-4.3 (light green), and the as-prepared TiO2-p-7.0 (dark green). A progressive rise in the intensity of oxo bond modes and a decrease in that of sulfate bonds can be observed in the spectra from brown to black and blue
The IR spectra of the as-prepared TiO2-a-4.3 (dark blue), TiO2-i-4.3 (light brown), and TiO2-w-4.3 (light green) showed progressively reduced sulfate stretches around 1111 cm−1, which was an indication of more complete hydrolysis of TiOSO4. Due to the very corrosive reaction media, we do not have in situ IR spectra available for this study; however, we speculate that the solvents played a role in the hydrolysis of TiOSO4. Given that the presence of sulfate in the products was an indication of incomplete hydrolysis, it was reasonable to conclude that acetic acid and isopropanol influenced the hydrolysis process. A slower and controlled sol‒gel process is preferred for a well-defined product and carboxylic acid and alcohols are often used for this purpose [45]. This is because carboxylic and ‒OR groups can coordinate with Ti(IV).
The spectrum of the as-prepared TiO2-p-7.0 in Fig. 1 (dark green) showed residual C‒H bands at 1360 cm−1, in addition to the oxo and OH bonds.
3.3 PXRD
The powder XRD patterns of TiO2-s, TiO2-a, TiO2-i, TiO2-w, and TiO2-p showed that anatase was already formed in the as-prepared samples without calcination (see the blue curves in Figs. 2 and 3 as examples), which makes this process attractive for certain potential applications. For example, it could be used for one-pot synthesis of organic‒inorganic hybrid composite materials, because it circumvents the calcination step that would destroy the organic components [46].

a The powder XRD patterns of TiO2-s-2.7-ap (as-prepared, dark blue), TiO2-s-2.7-400 (calcined at 400 °C, orange), TiO2-s-2.7-500 (gray), TiO2-s-2.7-600 (yellow), TiO2-s-2.7-700 (green), TiO2-s-2.7-800 (black), and TiO2-s-2.7-900 (light blue). b The powder XRD patterns of TiO2-w-4.3-ap (as-prepared, dark blue), TiO2-w-4.3-400 (calcined at 400 °C, orange), TiO2-w-4.3-500 (gray), TiO2-w-4.3-600 (yellow), TiO2-w-4.3-700 (green), TiO2-w-4.3-800 (black), and TiO2-w-4.3-900 (light blue). The peak indexes of anatase and rutile following PDF#00-021-1272 and PDF#21-1276, respectively

The powder XRD patterns of TiO2-p-2.2-ap (as-prepared, dark blue), TiO2-p-2.2-400 (calcined at 400 °C, orange), TiO2-p-2.2-500 (gray), TiO2-p-2.2-600 (yellow), TiO2-p-2.2-700 (green), TiO2-p-2.2-800 (black), and TiO2-p-2.2-900 (light blue). b = brookite; the peak indexes of anatase and rutile following PDF#00-021-1272 and PDF#21-1276, respectively
The anatase stabilities of these materials were different. The anatase in TiO2-p transitioned into rutile at a temperature as low as 500 °C, like many other TiO2 materials [47]. This low-temperature transition from anatase to rutile of TiO2-a likely followed the anatase‒brookite‒rutile route, given the fact that the brookite phase appeared in the as-prepared samples and after calcination at 400 °C too (see the orange and dark blue curves in Fig. 3). In contrast, the anatase phase of TiO2-s was kinetically stable up to 800 °C (Fig. 2). The stability may be ascribed to the small crystallite size of anatase [48], where higher temperatures would eventually cause sintering and loss of crystallite dispersion. At this point, it could be speculated that the high thermal stability of anatase in TiO2-s was due to the presence of sulfate, which was relatively thermal stable and potentially decelerated the sintering process. This speculation was also supported by a report that sulfate could facilitate anatase formation and stability [49]. However, this theory was challenged by the findings of TiO2-w-ap. As shown in Fig. 2b, the anatase phase in TiO2-w also exhibited high thermal stability; however, TiO2-w did not contain sulfate according to our IR and XPS analysis results.
It is noted that TiO2-i and TiO2-a samples are similar in kinetic stability when compared to TiO2-s. The anatase crystallite sizes of these samples will be described later.
3.4 TGA and XPS
We carried out both TGA and XPS analyses to understand why TiOSO4-derived TiO2 materials displayed better anatase stability than TiO2-p. According to Fig. 4a, there were three stages of significant weight loss for TiO2-s-2.7-ap. The first stage occurred before 350 °C, and the weight loss was attributed to the removal of both physical and chemical adsorbed water. The second (350‒436 °C) and the third stages (436‒800 °C) were mainly attributed to the loss of monodentate and bidentate sulfate, respectively. The XPS results of the same sample showed the presence of sulfur in the form of sulfate (Fig. 5a), and the molar ratio of S:Ti was as high as 1:4.8. After calcination at 700 °C, the S:Ti ratio dropped to 0.7:4, which agrees with the TGA weight loss in the same temperature range. It is noted that the decomposition of sulfate is a prolonged and complicated process, and a very high temperature is often required [50]. In contrast to TiO2-s, which showed weight loss at a temperature as high as 700 °C, TiO2-p did not have a significant weight loss above 400 °C. As shown in Fig. 4b, the first stage of weight loss at T < 125 °C was due to the removal of adsorbed water, and the second weight loss in the range of 190‒400 °C was assigned to the elimination of the ‒OC3H7 and hydroxyl groups. The residual hydrocarbon was detected by IR, as described earlier.

TGA plots of TiO2-s-2.7-ap (a) and TiO2-p-7.0-ap (b)

High-resolution XPS of TiO2-s-2.7-ap (left panel) and TiO2-s-2.7-700 (right panel) in the regions of S2p,1/2 (blue) and S2p,3/2 (magenta). The sulfur to Ti molar ratio was 1:4 for TiO2-s-2.7-ap and 0.07:4 for TiO2-s-2.7-700
In contrast to TiO2-p, which showed plateaued mass loss at 400–800 °C, TiO2-w and TiO2-i had a small and gradual weight loss above 400 °C (Fig. 6), which could be assigned to the loss of water and/or sulfate during sintering. The weight loss of TiO2-a above 400 °C was more pronounced, which was due to the higher concentration of sulfate in this sample (Table 3).

TGA plots of TiO2-w-4.3-ap (a), TiO2-i-4.3-ap (b), and TiO2-a-4.3-ap (c)
Nitrogen was noticeable in the TiO2 samples from the XPS results. Interestingly, there is still nitrogen after calcination at 700 and 900 °C (see TiO2-s-700 and TiO2-s-900 in Table 3). The presence of nitrogen was ascribed to the added ammonium hydroxide during the synthesis when ammonium could attach to sulfate to form bidentate ≡Ti‒SO4‒NH4 (Scheme 1). Upon calcination, ammonium cations decomposed to nitrogen atoms, and the latter was trapped in the TiO2 matrix to form doped TiO2 [51]. The doped nitrogen could be responsible for the higher stability of the anatase phase in these materials [52].

Ti-SO4-NH4 bidentate
Sulfate anions have been found to facilitate anatase formation from TiO62− octahedra and stabilize the phase [53,54,55,56]. This phenomenon is important for the application of TiO2 as a heterogenous catalyst when a high temperature and water content are involved [57].
It is noteworthy that XPS is a surface characterization technique, but it can still detect the top layers of atoms. The TiO2-s-2.7-ap sample was also extracted using NaOH, and the extraction solution was analyzed by means of ion chromatography (IC). The extracted sulfate was associated with the sample surface only. It is noted that this analysis technique has been widely used to analyze the sulfate poisoning of industrial catalysts. The IC result showed that the SO42− concentration was only 0.39 mole % (assuming that the sample contains TiO2 and sulfate), which was significantly lower than the XPS result (3.4%). This indicates that there was a significant amount of sulfate underneath the TiO2 surface.
The powder XRD data was also used to estimate the anatase lattice constants and unit cell volume, which could reveal if SO42− and N exist in the bulk TiO2 [58, 59]. As shown in Table 4, TiO2-s-2.7-900 has the same lattice constants and cell volume as the ideal anatase, suggesting all dopants were removed at this elevated temperature. On the other hand, all other samples (either as prepared or calcined at lower temperatures) have either different a, b, c lengths or unit cell volumes, indicating the doping effect of sulfate and N in these samples.
3.5 N2 physisorption
All TiO2 produced in this project exhibited Type IV isotherms, indicating that they were all mesoporous materials. Two types of isotherms are shown in Fig. 7: one for TiO2-s synthesized from TiOSO4 (black curve in Fig. 7a) and the other for TiO2-a synthesized from Ti(OPri)4 (orange curve in Fig. 7a). Both curves showed either H1 and/or H2 hysteresis loops in the relative pressure range of 0.4‒0.6, even though the black loop was less significant than the orange counterpart. In the high relative pressure range of 0.8‒0.95, the TiO2-s sample showed a significant secondary hysteresis loop. The interconnected pores generated these H2 loops, and the resulting pores may be attributed to the void space between the packed (or slightly fused) pseudo-spherical particles [60, 61]. The BJH pore size distributions of these materials are shown in Fig. 7b. While TiO2-a sample had only one maximum in the pore size distribution, TiO2-s showed two maxima (bimodal). The maxima at 3.3 and 3.6 nm from TiO2-s-2.7-400 and TiO2-a-2.2-400, respectively, were related to their hysteresis loops at 0.4‒0.6 relative pressure. These smaller mesopores were caused by the void space between the anatase crystallites, which were in the range of 7‒8 nm (see Table 5). The maximum at 37 nm (BJH desorption) corresponded to the void space between larger particles, about 100 nm (as shown in the SEM section), which were aggregated anatase crystallites. For H2 type hysteresis loops, the adsorption curve is normally used to estimate pore size, and the desorption curve is related to the pore neck size [62]. But for H1 type loops, the BJH desorption isotherm is preferred for the calculation of mesopore size distribution [63]. These large mesopores were related to the hysteresis loop in the 0.8‒0.95 relative pressure range. It is noted that the macropores contributed to the loops beyond 0.95 relative pressure, but the large pore sizes (>100 nm) cannot be accurately determined using the N2 physisorption technique.

a N2 physisorption isotherms of TiO2-s-2.7-400 (black) and TiO2-a-2.2-400 (orange); b Pore size distribution of TiO2-s-2.7-400 (black = BJH desorption, and gray = BJH adsorption) and TiO2-a-2.2-400 (brown = BJH desorption, and orange = BJH adsorption)
According to the N2 physisorption results, as shown in Table 5, the BET surface areas of TiO2-s-4.3 decreased from 203 to 141 and 100 m2 g−1 after heating to 400 and 500 °C, respectively. The surface area of this sample was still 10 m2 g−1 at 800 °C, which was comparable to the value of 11 m2 g−1 of TiO2 nanofibers calcined at 800 °C [64]. On the other hand, the surface areas of TiO2-p-2.2 dropped more dramatically after thermal treatment: its values were 209, 96, and 0.6 m2 g−1 at 80, 400, and 500 °C, respectively. The thermal stability of anatase crystallites can explain the significant difference between these calcined samples. The anatase crystallites of TiO2-s-4.3 did not transform but slowly grew from 6 to 37 nm when the calcination temperature increased from 80 to 800 °C (see Table 5 and Fig. 8a). The increment of crystallite size would decrease surface area (see the orange line in Fig. 8b) and cause smaller pores to collapse but generate larger pores (see the blue line in Fig. 8b). In contrast, the anatase crystals of TiO2-p-2.2 grew quickly from 8 (400 °C) to 30 nm (500 °C) and started to transform to rutile (Fig. 3). The phase change caused the surface area to drop significantly from 96 to 0.6 m2 g−1 when the temperature rose by 100 °C. Interestingly, the rutile crystallites of TiO2-p-2.2 did not grow, and their size remained at 41 nm when the temperature rose from 500 to 900 °C. It is also worth mentioning that the anatase phase of TiO2-s-2.7 was stable even though it grew up to 37 nm, which seemed to contradict the prediction that the anatase would convert to rutile when its crystallite size is over 11 nm [65]. As mentioned earlier, we believe that both doped nitrogen and sulfate would stabilize the anatase phase.

TiO2-s-4.3: a The plot of anatase crystallite size versus calcination temperature; b the plot of pore size versus the anatase crystallite size (blue) and the plot of BET surface area versus the anatase crystallite size (orange)
The N2 physisorption and powder XRD analysis results of the TiO2-i, TiO2-a, and TiO2-w series are summarized in Table 6. The BET surface area, pore size, and pore volume were similar among these samples, and they were also close to the values of the TiO2-s series. However, the pore size distributions of the TiO2-i, TiO2-a, and TiO2-w series are monomodal, in contrast to the bimodal TiO2-s series. This suggests that the solvents played a role in the microstructure of the sol‒gel product. At this point, however, it is unclear how acetic acid and isopropanol affected the aggregation of the anatase crystallites. These two solvents were added to chelate Ti(VI) to manipulate the product pore structure, but our IR results did not show any Ti-acetate bands in either the reaction intermediates or products.
3.6 SEM
The sol‒gel products of TiO2-s and TiO2-a exhibited monolithic structures and their dimensions were on the order of centimeters. The SEM images revealed their ubiquitous macropores on the surface of broken monoliths (Fig. 9a). The eruption of gas bubbles caused these macropores to form during the sol‒gel reactions. Both NH3 and propylene could have bubbled through the gel structure when the reaction temperature was at 65 °C. C3H6 was produced through the dehydration reaction of isopropanol (with Ti(IV) as a catalyst [66]), and it was believed to be more likely to generate the macropores [33]. These macropores are favorable for mass transfer for heterogeneous catalysis [67].

SEM images of TiO2-s-2.7-ap (a and b), TiO2-s-4.3-ap (c), and the sol‒gel product without the addition of water and NH4OH (d). The scale bar = 1 μm
Higher magnification SEM images revealed the spherical microstructures of TiO2. The diameters of these structures in TiO2-s-2.7-ap and TiO2-s-4.3-ap were about 100 and 40 nm, respectively. Because their anatase crystallite sizes were as small as 5.8 nm for TiO2-s-2.7-ap and 5.7 nm for TiO2-s-4.3-ap (see Table 5), each spherical particle in Fig. 9b, c should contain hundreds of anatase crystallites.
According to our IR observation, TiOSO4 reacted with HAc and IPA to a certain extent without adding H2O and NH4OH (see the black curve in Fig. 1). The SEM image, however, showed otherwise. Figure 9d shows the product of TiOSO4 reacting with HAc and IPA for 3 days at 65 °C. The rod-like morphology was identical to the raw material TiOSO4. It is noted that the starting material (67.3% TiOSO4) contained 25.5% water; apparently, this amount of water was not enough to complete the sol‒gel conversion.
The ubiquitous presence of the anatase phase in the TiO2 samples was confirmed by using HRTEM. In Fig. 10, the dominant (101) planes and some (004) planes of the anatase phase in the TiO2-s-2.7-400 sample can be observed.

HRTEM image of TiO2-s-2.7-400. The lattice spacing of 0.353 and 0.238 nm correspond to the (101) and (004) planes of anatase, respectively
4 Summary
Thermally stable anatase (kinetically stable) was synthesized by a low-temperature sol‒gel reaction of TiOSO4 with water in HAc and IPA. The anatase phase was readily produced without calcination and was stable at an elevated temperature as high as 800 °C, even though the crystallite size increased from ~5 to 37 nm. The improved thermal stability of anatase was attributed to a small amount of nitrogen and sulfate trapped in the TiO2 matrix. According to our TGA and XPS data, the nitrogen and sulfate contents in the anatase decreased with calcination temperature. The TiO2-s materials exhibited bimodal mesopores. The smaller mesopores (ca. 3 nm) were attributed to the void space between the anatase crystallites, and the large mesopores (10‒37 nm) were due to the void space between the TiO2 spherical particles that were an agglomeration of anatase crystallites. These TiO2 spherical particles were about 40 and 100 nm in diameter when the solution’s pH was 4.3 and 2.7, respectively. The TiO2 spherical particles were the building blocks of TiO2 xerogel monoliths in the order of centimeters. The SEM images revealed the presence of macropores with a dimension of a few micrometers. These macropores were generated by escaping gas bubbles of propylene and ammonia during the sol‒gel process. Our experimental results show that HAc and IPA played a role in the formation of large mesopores, even though the mechanism was not clear.
Because of the high thermal stability and large pore size of the resulting materials derived from TiOSO4, the anatase TiO2 would be useful for a variety of industrial applications. Our future work will be testing this material for both Claus catalysts for lower-temperature CS2 conversion and catalyst support for tail gas treatment. Our previous data showed that only excess sulfate would reduce the catalytic activities of Claus catalysts [68]. In the follow-up research, the effect of sulfate and N on the catalytic performance will be further studied.
Abbreviations
- IPA:
-
isopropanol alcohol
- IC:
-
Ion chromatography
- BET:
-
Brunauer Emmett Teller
- HAc:
-
acetic acid
- XRD:
-
X-ray diffraction
- PXRD:
-
powder X-ray diffraction
- XPS:
-
X-ray photoelectron spectroscopy
- SEM:
-
scanning electron microscopy
- IR:
-
infrared
- TGA:
-
thermogravimetric analysis
- IC:
-
ion chromatography
References
Hu W, Yang S, Yang S (2020) Surface modification of TiO2 for perovskite solar cells. Trends Chem 2(2):148–162. https://doi.org/10.1016/j.trechm.2019.11.002
Roose B, Pathak S, Steiner U (2015) Doping of TiO2 for sensitized solar cells. Chem Soc Rev 44(22):8326–8349. https://doi.org/10.1039/C5CS00352K
Schneider M, Baiker A (1997) Titania-based aerogels. Catal Today 35:339–365
Ni M, Leung MKH, Leung DYC, Sumathy K (2007) A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew Sustain Energy Rev 11(3):401–425. https://doi.org/10.1016/j.rser.2005.01.009
Kazemi ML, Sui R, Clark PD, Marriott RA (2019) Catalytic combustion of Claus tail gas: oxidation of sulfur species and CO using gold supported on lanthanide-modified TiO2. Appl Catal A: Gen 587:117256. https://doi.org/10.1016/j.apcata.2019.117256
Yanxin C, Yi J, Wenzhao L, Rongchao J, Shaozhen T, Wenbin H (1999) Adsorption and interaction of H2S/SO2 on TiO2. Catal Today 50(1):39–47. https://doi.org/10.1016/S0920-5861(98)00460-X
Chen D, Cheng Y, Zhou N, Chen P, Wang Y, Li K, Huo S, Cheng P, Peng P, Zhang R, Wang L, Liu H, Liu Y, Ruan R (2020) Photocatalytic degradation of organic pollutants using TiO2-based photocatalysts: a review. J Clean Prod 268:121725. https://doi.org/10.1016/j.jclepro.2020.121725
Rhodes C, Riddel SA, West J, Williams BP, Hutchings GJ (2000) The low-temperature hydrolysis of carbonyl sulfide and carbon disulfide: a review. Catal Today 59(3):443–464. https://doi.org/10.1016/S0920-5861(00)00309-6
Liu X, Zhang Q, Ye G, Li J, Li P, Zhou X, Keil FJ (2020) Deactivation and regeneration of Claus catalyst particles unraveled by pore network model. Chem Eng Sci 211:115305. https://doi.org/10.1016/j.ces.2019.115305
Tong S, Dalla Lana IG, Chuang KT (1997) Effect of catalyst shape on the hydrolysis of COS and CS2 in a simulated claus converter. Ind Eng Chem Res 36(10):4087–4093
Zhang W, Tian Y, He H, Xu L, Li W, Zhao D (2020) Recent advances in the synthesis of hierarchically mesoporous TiO2 materials for energy and environmental applications. Natl Sci Rev 7(11):1702–1725. https://doi.org/10.1093/nsr/nwaa021
Qi L, Cheng B, Yu J, Ho W (2016) High-surface area mesoporous Pt/TiO2 hollow chains for efficient formaldehyde decomposition at ambient temperature. J Hazard Mater 301:522–530. https://doi.org/10.1016/j.jhazmat.2015.09.026
Armstrong G, Armstrong AR, Canales J, Bruce PG (2005) Nanotubes with the TiO2 -B structure. Chem Commun 19:2454–2456
Rammal A, Brisach F, Henry M (2002) Hydrothermal synthesis of TiO2 anatase nanocrystals using hexaprismatic-shaped oxo-carboxylate complexes. Comptes Rendus Chim 5(1):59–66
Rotzinger FP, Kesselman-Truttmann JM, Hug SJ, Shklover V, Gratzel M (2004) Structure and vibrational spectrum of formate and acetate adsorbed from aqueous solution onto the TiO2 rutile (110) surface. J Phys Chem B 108(16):5004–5017
Kumar SG, Rao KSRK (2014) Polymorphic phase transition among the titania crystal structures using a solution-based approach: from precursor chemistry to nucleation process. Nanoscale 6(20):11574–11632. https://doi.org/10.1039/C4NR01657B
Rao BM, Roy SC (2014) Anatase TiO2 nanotube arrays with high temperature stability. RSC Adv 4(72):38133–38139. https://doi.org/10.1039/C4RA05882H
Gupta T, Samriti, Cho J, Prakash J (2021) Hydrothermal synthesis of TiO2 nanorods: formation chemistry, growth mechanism, and tailoring of surface properties for photocatalytic activities. Mater Today Chem 20:100428. https://doi.org/10.1016/j.mtchem.2021.100428
Chen C, Chen L, Chen S, Yu Y, Weng D, Mahmood A, Wang G, Wang J (2020) Preparation of underwater superoleophobic membranes via TiO2 electrostatic self-assembly for separation of stratified oil/water mixtures and emulsions. J Membr Sci 602:117976. https://doi.org/10.1016/j.memsci.2020.117976
Hashemizadeh I, Golovko VB, Choi J, Tsang DCW, Yip ACK (2018) Photocatalytic reduction of CO2 to hydrocarbons using bio-templated porous TiO2 architectures under UV and visible light. Chem Eng J 347:64–73. https://doi.org/10.1016/j.cej.2018.04.094
Gao Y, Yan N, Jiang C, Xu C, Yu S, Liang P, Zhang X, Liang S, Huang X (2020) Filtration-enhanced highly efficient photocatalytic degradation with a novel electrospun rGO@TiO2 nanofibrous membrane: implication for improving photocatalytic efficiency. Appl Catal B Environ 268:118737. https://doi.org/10.1016/j.apcatb.2020.118737
Siefering KL, Griffin GL (1990) Kinetics of low‐pressure chemical vapor deposition of TiO2 from titanium tetraisopropoxide. J Electrochem Soc 137(3):814–818. https://doi.org/10.1149/1.2086561
Wang W, Liu Y, Xue T, Li J, Chen D, Qi T (2015) Mechanism and kinetics of titanium hydrolysis in concentrated titanyl sulfate solution based on infrared and Raman spectra. Chem Eng Sci 134:196–204. https://doi.org/10.1016/j.ces.2015.05.006
Farjana SH, Huda N, Mahmud MAP, Lang C (2018) Towards sustainable TiO2 production: an investigation of environmental impacts of ilmenite and rutile processing routes in Australia. J Clean Prod 196:1016–1025. https://doi.org/10.1016/j.jclepro.2018.06.156
Sanchez C, Livage J, Henry M, Babonneau F (1988) Chemical modification of alkoxide precursors. J Non Cryst Solids 100:65–76
Vioux A (1997) Nonhydrolytic sol-gel routes to oxides. Chem Mater 9(11):2292
Colomer MT, Guzmán J, Moreno R (2010) Peptization of nanoparticulate titania sols prepared under different water–alkoxide molar ratios. J Am Ceram Soc 93(1):59–64. https://doi.org/10.1111/j.1551-2916.2009.03294.x
Mehranpour H, Askari M, Ghamsari MS, Farzalibeik H (2010) Study on the phase transformation kinetics of sol-gel drived nanoparticles. J Nanomaterials 2010:626978. https://doi.org/10.1155/2010/626978
Krivtsov I, Ilkaeva M, Avdin V, Amghouz Z, Khainakov SA, García JR, Díaz E, Ordóñez S (2015) Exceptional thermal stability of undoped anatase TiO2 photocatalysts prepared by a solvent-exchange method. RSC Adv 5(46):36634–36641. https://doi.org/10.1039/C5RA01114K
Wen M, Liu P, Xiao S, Mori K, Kuwahara Y, Yamashita H, Li H, Zhang D (2015) Uniform anatase single-crystal cubes with high thermal stability fully enclosed by active {010} and {001} facets. RSC Adv 5(15):11029–11035. https://doi.org/10.1039/C4RA16631K
Lv K, Xiang Q, Yu J (2011) Effect of calcination temperature on morphology and photocatalytic activity of anatase TiO2 nanosheets with exposed {001} facets. Appl Catal B: Environ 104(3):275–281. https://doi.org/10.1016/j.apcatb.2011.03.019
Sui R, Deering CE, Prinsloo R, Lavery CB, Chou N, Marriott RA (2020) Sol–gel synthesis of 2-dimensional TiO2: self-assembly of Ti–Oxoalkoxy–acetate complexes by carboxylate ligand directed condensation. Faraday Discuss. https://doi.org/10.1039/C9FD00108E
Sui R, Charpentier PA, Marriott RA (2020) Synthesizing 1D and 2D metal oxide nanostructures: using metal acetate complexes as building blocks. Nanoscale 12(35):17971–17981. https://doi.org/10.1039/D0NR03970E
Kozma K, Wang M, Molina PI, Martin NP, Feng Z, Nyman M (2019) The role of titanium-oxo clusters in the sulfate process for TiO2 production. Dalton Trans 48(29):11086–11093. https://doi.org/10.1039/C9DT01337G
Hixson AW, Plechner WW (1933) Hydrated titanium oxide. Thermal precipitation from titanium sulfate solutions. Ind Eng Chem 25(3):262–274
Ge L, Xu MX, Sun M (2006) Synthesis and characterization of TiO2 photocatalytic thin films prepared from refluxed PTA sols. Mater Lett 60(2):287–290. https://doi.org/10.1016/j.matlet.2005.08.036
Ngamta S, Boonprakob N, Wetchakun N, Ounnunkad K, Phanichphant S, Inceesungvorn B (2013) A facile synthesis of nanocrystalline anatase TiO2 from TiOSO4 aqueous solution. Mater Lett 105:76–79. https://doi.org/10.1016/j.matlet.2013.04.064
Štengl V, Bakardjieva S, Šubrt J, Szatmary L (2006) Titania aerogel prepared by low temperature supercritical drying. Microporous Mesoporous Mater 91(1):1–6. https://doi.org/10.1016/j.micromeso.2005.10.046
Kolen’ko YV, Burukhin AA, Churagulov BR, Oleynikov NN (2003) Synthesis of nanocrystalline TiO2 powders from aqueous TiOSO4 solutions under hydrothermal conditions. Mater Lett 57(5):1124–1129. https://doi.org/10.1016/S0167-577X(02)00943-6
Tian C, Zhang Z, Hou J, Luo N (2008) Surfactant/co-polymer template hydrothermal synthesis of thermally stable, mesoporous TiO2 from TiOSO4. Mater Lett 62(1):77–80. https://doi.org/10.1016/j.matlet.2007.04.092
Yang G, Ding H, Chen D, Ao W, Wang J, Hou X (2016) A simple route to synthesize mesoporous titania from TiOSO4: Influence of the synthesis conditions on the structural, pigments and photocatalytic properties. Appl Surf Sci 376:227–235. https://doi.org/10.1016/j.apsusc.2016.03.156
Sui R, Thangadurai V, Berlinguette CP (2008) Simple protocol for generating TiO2 nanofibers in organic media. Chem Mater 20(22):7022–7030. https://doi.org/10.1021/cm802091c
Jere GV, Patel CC (1962) Infrared absorption studies on peroxy titanium sulphate. Can J Chem 40(8):1576–1578. https://doi.org/10.1139/v62-238
Peak D, Ford RG, Sparks DL (1999) An in situ ATR-FTIR investigation of sulfate bonding mechanisms on goethite. J Colloid Interface Sci 218(1):289–299. https://doi.org/10.1006/jcis.1999.6405
Doeuff S, Henry C, Sanchez C, Livage J (1987) Hydrolysis of titanium alkoxides: modification of the molecular precursor by acetic acid. J Non Cryst Solids 8:206–216
Sui R, Charpentier PA, Marriott RA (2021) Metal oxide-related dendritic structures: self-assembly and applications for sensor, catalysis, energy conversion and beyond. Nanomaterials 11(7):1686
Hanaor DAH, Sorrell CC (2011) Review of the anatase to rutile phase transformation. J Mater Sci 46(4):855–874. https://doi.org/10.1007/s10853-010-5113-0
Zhang Q, Li C (2020) High temperature stable anatase phase titanium dioxide films synthesized by mist chemical vapor deposition. Nanomaterials 10(5). https://doi.org/10.3390/nano10050911.
Gao L, Zhang Q (2001) The promoting effect of sulfate ions on the nucleation of TiO2 (Anatase) nanocrystals. Mater Trans 42(8):1676–1680. https://doi.org/10.2320/matertrans.42.1676
Tagawa H (1984) Thermal decomposition temperatures of metal sulfates. Thermochim Acta 80(1):23–33. https://doi.org/10.1016/0040-6031(84)87181-6
Yuan J, Chen M, Shi J, Shangguan W (2006) Preparations and photocatalytic hydrogen evolution of N-doped TiO2 from urea and titanium tetrachloride. Int J Hydrog Energ 31(10):1326–1331. https://doi.org/10.1016/j.ijhydene.2005.11.016
Xu J-h, Dai W-L, Li J, Cao Y, Li H, He H, Fan K (2008) Simple fabrication of thermally stable apertured N-doped TiO2 microtubes as a highly efficient photocatalyst under visible light irradiation. Catal Commun 9(1):146–152. https://doi.org/10.1016/j.catcom.2007.05.043
Yan M, Chen F, Zhang J, Anpo M (2005) Preparation of controllable crystalline titania and study on the photocatalytic properties. J Phys Chem B 109(18):8673–8678. https://doi.org/10.1021/jp046087i
Bokhimi X, Morales A, Ortíz E, López T, Gómez R, Navarrete J (2004) Sulfate ions in titania polymorphs. J Sol Gel Sci Technol 29(1):31–40. https://doi.org/10.1023/B:JSST.0000016135.02238.0e
Oliveri G, Ramis G, Busca G, Escribano VS (1993) Thermal stability of vanadia–titania catalysts. J Mater Chem 3(12):1239–1249. https://doi.org/10.1039/JM9930301239
Colón G, Sánchez-España JM, Hidalgo MC, Navío JA (2006) Effect of TiO2 acidic pre-treatment on the photocatalytic properties for phenol degradation. J Photochem Photobiol A Chem 179(1):20–27. https://doi.org/10.1016/j.jphotochem.2005.07.007
Sadighi S, Mohaddecy SRS, Rashidzadeh M, Nouriasl P (2020) Kinetic-based models for alumina and titania claus catalysts based on experimental data. Pet Chem 60(3):321–328. https://doi.org/10.1134/S0965544120030196
Kryukova GN, Zenkovets GA, Shutilov AA, Wilde M, Günther K, Fassler D, Richter K (2007) Structural peculiarities of TiO2 and Pt/TiO2 catalysts for the photocatalytic oxidation of aqueous solution of Acid Orange 7 Dye upon ultraviolet light. Appl Catal B Environ 71(3):169–176. https://doi.org/10.1016/j.apcatb.2006.06.025
Mohamed MM, Bayoumy WA, Khairy M, Mousa MA (2006) Synthesis and structural characterization of TiO2 and V2O5/TiO2 nanoparticles assembled by the anionic surfactant sodium dodecyl sulfate. Microporous Mesoporous Mater 97(1):66–77. https://doi.org/10.1016/j.micromeso.2006.07.028
Lim S-Y (2019) Bimodal porous structure tin oxide anode materials for lithium ion batteries. J Ind Eng Chem 78:284–294. https://doi.org/10.1016/j.jiec.2019.06.001
Müllner T, Unger KK, Tallarek U (2016) Characterization of microscopic disorder in reconstructed porous materials and assessment of mass transport-relevant structural descriptors. N J Chem 40(5):3993–4015. https://doi.org/10.1039/C5NJ03346B
Atwa M, Li X, O’Connell D, Sui R, Marriott R, Birss V (2023) Nanoengineering of pore neck size within ordered nanoporous carbon powders: improving ion transport rates for clean energy applications. Chem Mater 35(2):395–404. https://doi.org/10.1021/acs.chemmater.2c02069
Sing KSW, Williams RT (2004) Physisorption hysteresis loops and the characterization of nanoporous materials. Adsorpt Sci Technol 22(10):773–782. https://doi.org/10.1260/0263617053499032
Sui R, Carefoot SK, Lavery CB, Deering CE, Lesage KL, Chou N, Rose CJ, Marriott RA (2017) Organosulfur adsorbents by self-assembly of titania based ternary metal oxide nanofibers. J Mater Chem A 5(20):9561–9571
Zhang H, Banfield JF (2000) Understanding polymorphic phase transformation behavior during growth of nanocrystalline aggregates: insights from TiO2. J Phys Chem B 104:3481–3487
Schubert U (2005) Chemical modification of titanium alkoxides for sol-gel processing. J Mater Chem 15:3701–3715
Takahashi R, Sato S, Sodesawa T, Arai K, Yabuki M (2005) Effect of diffusion in catalytic dehydration of alcohol over silica–alumina with continuous macropores. J Catal 229(1):24–29. https://doi.org/10.1016/j.jcat.2004.10.010
Sui R, Lavery CB, Deering CE, Prinsloo R, Li D, Chou N, Lesage KL, Marriott RA (2020) Improved carbon disulfide conversion: Modification of an alumina Claus catalyst by deposition of transition metal oxides. Appl Catal A Gen 604:117773. https://doi.org/10.1016/j.apcata.2020.117773
Acknowledgements
The authors are grateful to NSERC for a Discovery Grant and the supporting member companies of ASRL. We thank Dr. Mark Biesinger of Surface Science Western, Western University for the XPS analysis. We also thank Drs. Christopher Debuhr, Yongliang Wang, and Robert Marr of the University of Calgary for their SEM, TEM, and powder XRD work.
Funding
This research has been funded through the Natural Science and Engineering Research Council of Canada (NSERC) and Alberta Sulfur Research Ltd. (ASRL) Industrial Research Chair program in Applied Sulfur Chemistry.
Author information
Authors and Affiliations
Contributions
The manuscript was written with the contributions of all authors. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sui, R., Jacobs, J.H., Chou, N. et al. Facile synthesis of thermally stable anatase titania with a high-surface area and tailored pore sizes. J Sol-Gel Sci Technol 107, 289–301 (2023). https://doi.org/10.1007/s10971-023-06117-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-023-06117-7