
Abstract
Instrumental neutron activation analysis (INAA) relies on constant neutron flux densities throughout the activated samples. Although this concept is true for most typical samples, occasionally, the presence of highly neutron absorbing nuclides in the sample may cause a neutron flux density suppression which would ultimately lead to distorted results in the INAA. Here, we have investigated artificial samples with a high manganese (Mn) content. By adding aqueous gold solution, we introduced a liquid in-situ neutron flux monitor into the sample. An Mn content ≤ 50% shows little effect to the internal neutron flux density, however, the flux can be suppressed by ca. 20% when the Mn content reaches 63.2%.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The principle of instrumental neutron activation analysis (INAA) relies on the fact that, once a sample is irradiated with neutrons, a small but representative number of isotopes of each element in the sample will be activated in a variety of nuclear reactions. The activity induced by these nuclear reactions will directly correspond to the amount of the respective element in the sample [1]. The probability for a nuclear reaction to take place is expressed as the nuclear cross section (σ). In case of a INAA, the cross section typically determines the probability for neutron capture in a stable nucleus, leading to an (n,γ) reaction and thus yielding an unstable nucleus which decays with a certain half-life. Upon decay, the resulting radiation can be measured and acts as the analytical signal in INAA.
A fundamental principle of INAA, however, is that the neutron flux density is regarded as constant throughout the sample, with only marginal losses due to neutron capture. Neutron flux suppression inside an irradiated sample can be calculated using Eq. (1), where Qx is the neutron flux density after penetration through the sample, Qx0 is the initial neutron flux density, NA is the number of target atoms, and s is the thickness of the sample.
Samples with high neutron absorbing properties, therefore, limit the applicability of INAA [2, 3], because this fundamental precondition of a constant neutron flux density throughout the sample may not be fulfilled any longer and, even worse, no analytical signal will be generated indicating the problematic neutron flux suppression that has taken place [4].
In our previous work on neutron absorption activation analysis (INAAA) [5], we have introduced a simple method that allows quantification of neutron absorbers in a sample by adding an activatable tracer to the sample and to experimentally determine the neutron flux suppression via the indicator’s activation product’s activity. In the present study, we investigate the applicability of this principle for samples with high manganese (Mn) content. Mn is considered a highly suitable element for INAA, however, in typical geological [6], biological [7], food [8] etc. samples, the Mn content is not considered high enough to cause any problematic neutron flux density suppression. Here, we want to lay the foundations of analytical quality control by applying INAAA principles in artificial samples with high Mn content. Manganese is a monoisotopic element, with a high cross section for the 55Mn(n,γ)56Mn reaction with thermal neutrons (σ = 13.36 b), and a short-lived activation product 56Mn (T1/2 = 2.58 h) that emits several γ photons with high intensities (847, 1811, 2113 keV, and others). By soaking the Mn-containing sample with Au3+ solution as an indicator, we attempt to quantify the neutron flux suppression via the activity of the activation product 198Au after the irradiation.
Experimental
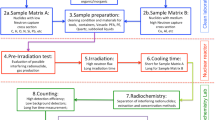
To quantify the possible suppression of the neutron flux inside the samples, various manganese containing chemicals (MnCO3 (Sigma Aldrich, p.a.) and MnO2 (Merck, p.a.) were used. These chemicals were diluted with aluminum oxide (Al2O3, Merck, p.a.) as a “neutron inert” filler to adjust the effective Mn content of the mixture. The chemicals were weighed into polyethylene (PE) vials, which could be sealed after preparation to prevent any kind of contamination and which allowed for re-opening if necessary. These PE vials (Kartell LABWARE) are cone-shaped at their bottom and then become cylindrical closer to the top. Due to uncertainties from the manufacturing process of the PE vials, they exhibit a maximum diameter of 0.42 cm and a maximum filling level of 3.8 cm. In the experiments conducted in this study, the filling level in the vial varied between 1.8 and 2.3 cm depending on the density of the mixture as a result of the variable Mn contents. The effective Mn content in these artificial samples ranged between 6.3 and 63.2%.
The indicator in this study was 197Au in the form of HAuCl4 solution. A CentriPUR standard Au3+ solution was then pipetted and soaked into the mixture, yielding the activity of 198Au as the analytical signal of the neutron activation, or the lack thereof as a signal of the neutron flux density suppression, respectively. Naturally, the distribution between the powdered mixture and the Au-containing solution was heterogeneous, but since the powder was finely ground, we assumed an even distribution throughout the mixture. The activation product 198Au has a high cross section for thermal neutron capture (σ = 98.65 b). The activation product 198Au (T1/2 = 2.69 d) emits one dominant γ photon at 412 keV.
The amount of Au3+ solution needed was determined visually by observing the amount of solution that would be needed to soak the entire solid mixture with solution but avoid any excess of solution that would no longer be in contact with the Mn-containing solid matrix. Subsequently, the vials were manually shaken and centrifuged. In case any residual solution was observed after this step, the vial was discarded and the procedure was repeated by preparing a new mixture. After finding the right amount of solution to be pipetted and weighed into the mixture, quartz sand (SiO2, Merck, p.a.) was used to fill up the vials to limit the mixture’s ability to move inside the vial due to vibrations and shock during transport in the pneumatic sample transfer system. The approach of visually determining the apt amount of Au3+ solution introduces some uncertainty because every other parameter was determined precisely by weighing on a scale with a well-known and well-defined uncertainty of measurement. A certain amount of excess liquid, however, cannot be ruled out using this visual technique. The used amounts of Mn mixture and Au3+ solution were 0.0941 ± 0.0019 g of solution soaked into 0.14000 ± 0.00001 g of MnCO3 mixture. The weighed in masses for the more important MnO2 samples can be found in Table 1.
The vials were labeled, rinsed and decontaminated with distilled water in an ultrasonic bath before being transferred to an irradiation capsule for the rabbit system. The capsules were then closed, labeled, and irradiated for 120 s in a TRIGA Mk-II reactor at a neutron flux density (Φ = 3⋅1012 cm−2 s−1) using the automatic pneumatic transport system of the TRIGA Center Atominstitut of TU Wien. Since all of the capsules were irradiated for the same amount of time, the duration of transport in the system is deemed to cancel out for all the capsules and does not require any further consideration. The dose rate after irradiation was considerable (400 µSv/h in 60 cm distance). Hence, the capsules were stored inside a lead shielding for about 24 h after removal from the rabbit system. This cooling time allowed for a substantial cooling of both 28Al (T1/2 = 2.25 min) and 56Mn, yet allowing for sufficient activities of 198Au for measurement.
The cooling times were noted for each indiviual for decay correction. Measurement was performed on a 222cm3 HPGe detector (1.78 keV resolution at the 1332 keV 60Co peak; 48.2% relative efficiency) [9] with a preloaded filter and loss-free counting system.
Results and discussion
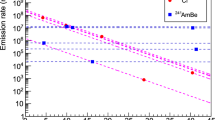
Figure 1 shows the neutron flux suppression that occurs over different manganese concentrations in the MnCO3 and MnO2 mixtures, as determined via the 198Au activity. Initially, MnCO3 was selected as the go-to chemical as it contains no other neutron-absorbing nuclei but Mn (neutron absorption by C and O are regarded as negligible) and the Mn content in MnCO3 (47.8%) was deemed to resemble a “typical Mn-rich sample” quite realistically. At a later stage, MnO2 was included due to its chemical properties (resistance against acids). In any case, the mixtures were prepared from 10 to 100% share of the respective Mn chemical (residual percentages are Al2O3) and the real manganese content was calculated accordingly. Experiments including two different species of MnCO3 are plotted: One, relatively coarse,”normal” grained MnCO3 and two, finely grained MnCO3 after grinding the powder in an agate mortar. Indeed, some deviations were observed, with the fine-grained MnCO3 showing higher neutron flux suppression than the normal-grained MnCO3. This observation does not come entirely unexpected, as one can assume that the coarser grained material exhibits a lower degree of homogeneity within the sample, thus allowing the 197Au indicator to be exposed to more neutrons with less interference by surrounding Mn nuclei. This specific difference in neutron flux density can be quantified at around five to ten percent. The fluctuation between the data points may be explained by individual inhomogeneities within the samples. Lastly, one has to consider that MnCO3 slightly reacted with the acidic HAuCl4 solution causing the formation of tiny bubbles of CO2 within the matrix. The data points for the MnO2 mixtures hence unsurprisingly indicate a steadier suppression of the neutron flux density. Following this series, another series of MnO2 mixtures with a manganese content of 0%, 10%, 20%, 50%, and 63.2% (the latter representing pure MnO2) were prepared under identical irradiation conditions. For a better statistical significance, each mixture was prepared and irradiated in five replicates, and the mean values were calculated. The results are shown in Fig. 2. The average weighed in masses that were used to prepare the MnO2/Al2O3 mixtures are presented in Table 2. Here, the error bars in y-dimension are visible and the error bars in x-dimension are also given, but smaller than the data points. The error bars in y-dimension represent the 1σ-environment. The error bars in x-dimension range around 10–7 due to the weighing uncertainties. Therefore, the error bars in x-dimension represent the 3σ environment around the mean value. Figure 2 shows the neutron flux density suppression as a function of the manganese content in MnO2/Al2O3 mixtures. It can be seen that the neutron flux density is not suppressed significantly until a manganese content of 50% in the sample is exceeded. The data point at 0% of manganese content represents a vial that was just filled with pure Al2O3 powder (soaked with Au3+ indicator). This data point is therefore to be regarded as a mathematical reference point and has no error bars by definition, although, naturally, fluctuating counting statistics for the 412 keV peak was observed. If the 1σ environment around the data points up until 50% is considered, the data points for 5%, 10%, 20%, and 50% show a virtually identical neutron flux density, before a significant decrease can be observed when irradiating the pure MnO2 mixtures. Table 3 shows the theoretical effective neutron flux density that was calculated for each manganese mixture according to Eq. (1) vs. the empirically determined values for the effective neutron flux density for the same mixtures as graphically presented in Fig. 2. A discrepancy of up to 20% can be identified when comparing the data pairs. This high deviation cannot be explained neither with fluctuations in the reactor’s neutron flux density, nor the uncertainty introduced by the analytical scale. Potential factors impacting the deviation between theoretical and empirical neutron flux density values include a lack of homogeneity due to the manual preparation of the mixtures as well as the impact from epithermal and fast neutrons which are not accounted for in the theoretical effective neutron flux density values.

Effective neutron flux density suppression in MnO2 and MnCO3 samples with different manganese concentrations

Effective neutron flux density observed in MnO2 samples with different manganese concentrations
The disparity between calculated and experimental effective neutron flux densities illustrates that an experimental validation is still crucial in analytical quality control, as not all factors can be accounted for in a theoretical assessment. Most importantly, the study shows that for a Mn content of ≤ 50%, INAA can be applied without major concerns regarding internal neutron flux suppression inside the sample (when using standard vials).
Although presumably less relevant in our case, gamma ray self-attenuation of the activation products’ radiation may become a problem when (a) inhomogeneity issues with respect to atomic numbers of constituents arise within the sample, and/or (b) the gamma-energy of the indicator nuclide is so low that self-attenuation becomes more problematic [10, 11].
Conclusions
This work is dedicated to the investigation of the potential effects of highly neutron-absorbing nuclei in “non-typical” materials for INAA. Nuclides with a high cross-section may cause an internal neutron flux suppression and hence thwart the generally accepted approach in INAA that assumes that, for most types of samples, the neutron flux density remains constant throughout the sample. By adding aqueous gold solution to the powdered sample, we introduce an indicator of the effective neutron flux density inside the sample. This allows for the observation of the actual conditions during neutron irradiation via measurement of the activity of the activation product 198Au.
Here, we have focused on 55Mn as a major matrix constituent in non-typical samples. We can demonstrate experimentally, that for samples with a Mn content of ≤ 50%, no adverse effects are anticipated due to effective neutron flux suppression inside the sample. Above 50%, manganese may cause ca. 20% neutron flux suppression.
This study can also act as a blueprint for analytical quality control protocols with other highly neutron absorbing properties, as the analytical approach using the addition of an activatable indicator can be expanded to other matrices as well as other indicator materials.
In the present case, a detailed INAA-analytical protocol for the analysis of samples that are suspected of containing manganese of uncertain (but high) concentration has been presented. By preparing mixtures of MnO2 with Al2O3 as an “inert filler,” a calibration line could be established that proves that neutron self-absorption inside the sample may become problematic, even when theoretical considerations might pretend a false sense of security.
We could show that, while 50% Mn content is not yet, problematic, 63.2% is. For future investigations, the range of Mn content between 50% and 63.2% appears as an attractive area of research for neutron self-absorption studies.
Data availability
All data reported and discussed in this study are available either in the main text or in the Supplementary Information.
References
Hevesy G, Levi H (1936) Action of slow neutrons on rare earth elements. Nature 137:185. https://doi.org/10.1038/137185a0
Blaauw M (1996) The derivation and proper use of stewart’s formula for thermal neutron self-shielding in scattering media. Nucl Sci Eng 124:431–435. https://doi.org/10.13182/NSE96-A17921
Lindstrom RM, Fleming RF (2008) Neutron self-shielding factors for simple geometries, revisited. Chem Anal (Warsaw) 53:855–859
Greenberg RR, Bode P, De Nadai Fernandes EA (2011) Neutron activation analysis: A primary method of measurement. Spectrochim Acta, Part B 66:193–241. https://doi.org/10.1016/j.sab.2010.12.011
Steinhauser G, Merz S, Sterba JH (2013) Instrumental neutron absorption activation analysis. J Radioanal Nucl Chem 296:165–168. https://doi.org/10.1007/s10967-012-1925-5
Steinhauser G, Sterba JH, Bichler M (2007) “Chemical fingerprints” of pumice from Cappadocia (Turkey) and Kos (Greece) for archaeological applications. Appl Radiat Isot 65:488–503. https://doi.org/10.1016/j.apradiso.2006.12.004
Adlassnig W, Steinhauser G, Peroutka M, Musilek A, Sterba JH, Lichtscheidl IK, Bichler M (2009) Expanding the menu for carnivorous plants: Uptake of potassium, iron and manganese by carnivorous pitcher plants. Appl Radiat Isot 67:2117–2122
De Nadai Fernandes EA, Furlan GN, Lima RC, Bacchi MA, Sarriés SRV, Sarriés GA (2022) Elemental profile of dietary supplements and agricultural byproducts evaluated by neutron activation analysis. J Radioanal Nucl Chem 331:5487–5492. https://doi.org/10.1007/s10967-022-08662-x
Steinhauser G, Bichler M (2008) Adsorption of ions onto high silica volcanic glass. Appl Radiat Isot 66:1–8. https://doi.org/10.1016/j.apradiso.2007.07.010
Fleming RF (1982) Neutron self-shielding factors for simple geometrics. Int J Appl Radiat Isot 33:1263–1268. https://doi.org/10.1016/0020-708X(82)90247-2
Debertin K, Helmer RG (1988) Gamma- and X-Ray spectrometry with semiconductor detectors. North Holland, Amsterdam
Acknowledgements
We would to thank the reactor operating team at TRIGA Center Atominstitut for their support in the irradiation procedures, as well as the TRIGA Center’s radiation protection staff for their invaluable help in dealing with highly activated samples. We thank Michaela Foster, Johannes H. Sterba, and Jan M. Welch for their continued support in this study.
Funding
Open access funding provided by TU Wien (TUW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schramm, LM., Nemetz, M., Renz, F. et al. Experimental validation of the neutron flux suppression in samples with high manganese content in instrumental neutron activation analysis. J Radioanal Nucl Chem (2024). https://doi.org/10.1007/s10967-024-09671-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10967-024-09671-8