
Abstract
In radiochemical analysis of decommissioning waste, reference materials are not commercially available and consequently intercomparison exercises are needed for method validations. This paper reports the results from an intercomparison exercise focusing on analyses of α-emitter radionuclides in spent ion exchange resin. The exercise and statistical analysis of the submitted results were carried out based on ISO 13528 standard according to which the assigned values were calculated from the participant’s results and the performances were assessed using z-scores, which showed excellent performance for all 241Am analyses, good performance for 238Pu analyses, tolerable performance for 239,240Pu analyses, and questionable 243,244Cm analyses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Characterisation of radioactive decommissioning waste may be divided into (i) easy to measure (ETM), and (ii) difficult to measure (DTM) radionuclides. The ETMs are analysed using γ-spectrometry, which is a non-destructive characterisation method for γ-emitters. The DTMs, which may be further divided to α- and β-emitters, require destructive radiochemical characterisation methods which include separation of the DTMs from the matrix and from other interfering radionuclides or elements. Even though α- and β-emitting radionuclides are radiochemically separated and purified using similar methods (including ion exchange chromatography, extraction chromatography and precipitation), the measurement techniques generally are α-spectrometry for α-emitters and liquid scintillation counting (LSC) for β-emitters. The measurement source in α-spectrometry is ideally a very thin layer of the purified DTM fraction on a smooth surface. Preparation of the measurement source can be carried out using micro-coprecipitation on a filter or electrodeposition on a metal disc. The measurement source is measured in vacuum at a set distance from the detector. The thin layer minimises self-absorption of α-particles and consequently the α-spectrum can exhibit discrete energy specific peaks. Widening of the peaks is linked with self-absorption of the α-particles and the limitations of the detector system that lowers the resolution of the measured α-spectrum and can make the interpretation of the spectrum more complex. In LSC measurements of β-emitters, an aliquot of purified fraction is mixed with a scintillation cocktail prior to measurement. Ideally, the chemical and colour quenching from the separated material should be minimised. In addition to α-spectrometry and LSC measurements, measurement of DTMs can be carried out using mass spectrometric techniques, such as inductively coupled plasma mass spectrometry (ICP-MS); both techniques provide isotopic information on the sample and ICP-MS may provide information about the sample that is inaccessible by radiometric techniques, such as the determination of 239Pu and 240Pu. The mass spectrometric analyses are challenged by isobaric and polyatomic interferences from isotopes and molecules with similar masses, complicating the determination of DTM content in the sample. However, significantly lower detection limits at µBq levels may be reached using ICP-MS compared to LSC, which detection limits are typically at the mBq level [1].
Characterisation of decommissioning waste relies on application of material specific scaling factors which link a key ETM (e.g., 60Co in steel) to DTMs (e.g., 55Fe and 63Ni in steel). Scaling factors are determined preferably before dismantling to measure only the ETMs and calculate the DTMs when large number of waste packages are characterised during dismantling. As with any analytical method, method validation is required for reliable results and to demonstrate confidence in such results. Commercial reference materials can be analysed using the analytical method and satisfactory results strongly support validation of the method. Participation in intercomparison exercises supports method validation.
Four years of intercomparison exercises within the Nordic Nuclear Safety Research (NKS) community on radiochemical analysis of DTM radionuclides in decommissioning waste were organised between 2019 and 2022 [2,3,4,5]. The capabilities of the participants from Nordic and non-Nordic countries ranged from status of building up the capabilities to advanced laboratories with routine analysis services. The studied materials were activated steel [2, 6], activated concrete [3, 7] and spent ion exchange resin [4, 5, 8]. The exercises were organised, and the results were statistically analysed according to the requirements of the ISO 13528 standard [9]. The coordinator carried out the homogeneity studies of the materials used with γ-spectrometric analysis of a key ETM, which consistently showed < 2% relative standard deviation (RSD) [2,3,4,5]. Additionally, the homogeneities of individual materials were calculated and confirmed according to the ISO 13528 standard.
Individual partners had several months to carry out the analyses; discussions on the challenges and exchange of information were encouraged. The coordinator collected the analysis results calculated on a reference date and organised a preliminary meeting, in which the preliminary results were discussed and possible reasons for deviations from the general trends were suggested. Where a result seemed to be a clear outlier (e.g., an order of magnitude difference to the general trend), the partner was contacted in advance and advised to check the results for possible calculation errors and similar errors, referred to as ‘blunders’ in ISO 13528. After the preliminary meeting, each partner had about a month to re-check their results (e.g., update of quenching curves) and submit the final results. The project reports include both the preliminary and final results [2,3,4,5] whereas the peer-reviewed articles include the final results with more detailed statistical analysis and critical considerations regarding the analysis methods and radionuclides [6,7,8].
This paper reports the analysis of α-emitting DTM nuclides in the spent ion exchange resin, which β-emitting DTM nuclides and ETMs were studied in the previous project [5]. Nine laboratories participated in the exercise named RESINA. The main α-emitting DTM nuclides analysed were 241Am, 234U, 235U, 238U, 238Pu, 239,240Pu, 242Cm, and 243,244Cm with gross α-activity measurements and 241Pu (β-emitter) as optional analyses. Additionally, preliminary 237Np results were also submitted but not reported in the final results as measurement is compromised by the lack of a suitable yield tracer [5]. Statistical analyses according to the ISO 13528 standard were carried out for sets of results for which at least five above limit of detection results (one result per partner) were submitted. Additionally, an example of an extended data analysis is shown for 241Am results.
Sample history, homogeneity and stability
The spent ion exchange resin, which had already been studied in the previous intercomparison exercise named DTM Decom III [4], was a nuclear grade FINEX C/A 850 H mixed exchanger with 1:1 ratio of cation and anion exchange capacity. The resin particle sized was between 10 to 200 µm. The resin had been used for cleaning of a primary circuit of a nuclear power plant and it was relatively fresh as short-lived radionuclides, such as 54Mn, were detected [4]. Homogeneity and stability of the samples were established in the DTM Decom III project according to the ISO 13528 standard [4].
Origin of α-emitting DTM nuclides in spent ion exchange resin
The ETMs and DTMs in the primary circuit of a nuclear reactor are divided into fission products, coolant activation products, and corrosion products [10]. The ETMs and DTMs in the coolant are removed from circulation via cleaning loops with ion exchange resins. The α-emitters originate from the nuclear fuel. For example, a light water reactor spent fuel with a burnup of 50 GWd/tHM consists of approximately 93.4% uranium (approximately 0.8% 235U), 5.2% fission products, 1.2% plutonium, and 0.2% minor transuranium elements, mainly neptunium, americium and curium [11]. Although the fuel is in the form of low solubility UO2 and the fuel cladding is relatively inert, some activity does originate from the fuel, although most of the radionuclides in the primary circuit originates from corrosion and consequently there are overwhelmingly more β-emitting radionuclides than α-emitters in the spent ion exchange resin.
Methodology for statistical analysis
The intercomparison exercises and statistical analyses in the DTM Decom I-III and RESINA projects were performed according to the ISO 13528 standard on proficiency testing by interlaboratory comparison [9]. As noted before, the spent ion exchange resin came from an operating nuclear power plant and thus the reference values of α-emitting DTM nuclides were not known in advance. Therefore, the assigned values, against which the performances were assessed, were calculated from the reported results as recommended in the ISO 13528 [9]. The assigned values, robust mean values and robust standard deviations, were calculated using Algorithm A, which is robust for outliers up to 20% [9]. The iterations of the robust mean and standard deviations were carried out until there was no change in the third significant figure [9]. The performance assessments were carried out using z-score of Eq. (1), which is the recommended method when the assigned value is calculated from the submitted results [9]. When the robust standard deviation was large (> 20%), the uncertainty of the assigned value from equation was used as σpt [9]. The submitted results (xi) were assessed against the assigned values derived from the participants’ results [9]. The z-score results were acceptable when |z|≤ 2.0, a warning signal was given for results with 2.0 <|z|< 3.0, and |z|≥ 3.0 results were unacceptable [9].
where.
\({x}_{\mathrm{pt}}\):the assigned value.
\({\sigma }_{\mathrm{pt}}\):standard deviation for the proficiency assessment
where.
\({s}^{*}\):robust standard deviation of the results.
\(p\):number of samples
In addition to the previous exercises, an extended data analysis was carried out for the 241Am results. If the dataset is large enough, the data analysis may be extended, and outliers from the dataset identified using the Pierce criterion [12, 13]; note that this does not necessarily establish an assigned value, although it may be used to do so if the intercomparison exercise organiser is confident that there are no systematic biases within the dataset. Using the culled dataset allows the establishment of \({\sigma }_{\mathrm{pt}}\), based on the results submitted by participants. Additionally, the quality of reported uncertainties may be established through means of an \(R\)-score which is calculated using Eq. (3).
The \(R\)-test is passed if \({R}_{\mathrm{lower}}<{R}_{i}<{R}_{\mathrm{upper}}\).
Having set the pass fail criteria for the ζ-score, z-score and relative uncertainty tests, the outcomes may be combined [14] and the dataset may be analysed as follows:
-
Results with no reported uncertainty are rejected.
-
The \(\zeta\)-score is calculated and \(\zeta\)-scores \(>5.026\) (a different limit may be used) are flagged as improbable deviations, where:
$$\zeta =\frac{\left|{x}_{i} -{x}_{\mathrm{pt}}\right|}{\sqrt{{u}^{2}\left({x}_{i}\right)+{u}^{2}\left({x}_{\mathrm{pt}}\right)}}$$ -
The \(z\)-score is calculated as usual, where \({\sigma }_{p}=A\bullet {u}_{\mathrm{rel}}\left(\mathrm{med}\right)\) and \({u}_{\mathrm{rel}}\left(\mathrm{med}\right)\) is determined after rejection of outliers according to the Pierce criterion.
-
Finally, the \(R\)-score is calculated as above.
The outcome from the three tests may be interpreted as explained in Table 1. The combination of these tests allows the participant result to be assessed against the assigned value, and comparison with the general performance of the participants in the intercomparison exercise.
Experimental
The radiochemical analysis methods for α-emitting DTM nuclides included utilisation of acid digestion or leaching, precipitations, and ion exchange and chromatographic column separations prior to measurement of the separated and purified actinide fractions (Table 2). In previous exercises, the β-emitting DTM nuclides were mainly analysed using LSC and in one case using ICP-MS (90Sr in spent ion exchange resin) [4]. However, in this exercise four measurement techniques were implemented.
-
α-spectrometry for the measurement of gross α-activity measurements, 241Am, uranium, plutonium, curium isotopes,
-
γ-spectrometry for measurement of 241Am and 235U,
-
mass spectrometry for measurement of uranium isotopes, and
-
LSC for the measurement of gross α-activity and 241Pu.
The detailed account of preliminary (replicate results included) and final results are presented by Leskinen et al. [4] whereas general descriptions are given in following sub-chapters and Table 2. The statistical analysis of the final results was carried out when at least 5 above limit of detection data entries per radionuclide and only one data entry per sample (no replicate results) were available.
Overview of the radiochemical methods
The published radiochemical methods [15,16,17,18,19,20,21,22,23,24,25,26,27,28] used for the α-emitting DTM nuclide analyses were presented by Leskinen et al. [5]. Resin samples from 10 to 250 mg were acid digested in different mixtures of acids (e.g., HNO3, HCl, H2SO4) and oxidising agents (e.g., H2O2, Fenton’s reagent) with addition of heat on a hotplate, microwave, sand bath or under reflux. Pre-treatment with drying at 150 °C and ashing at 500 °C was carried out for Sample 4. Radioactive tracers were used in yield determinations in all Samples (Samples 2–10) and additionally carriers and hold-back carriers, such as Fe, Co, Ni, Eu, Cs, Sb and Sr, were added in Samples 2 and 8. The separation techniques included precipitation including actinide coprecipitation with iron hydroxide and lanthanum coprecipitation in ammonia, the use of ion exchange resin (AG1 × 8®) and extraction chromatography media (TEVA®, TRU®, UTEVA® and DGA®). The purified fractions contained:
-
uranium isotopes,
-
plutonium isotopes and possibly neptunium, and
-
241Am and curium isotopes.
The measurement sources for the α-spectrometry analyses were prepared using precipitation (Samples 2 and 5) and electrodeposition (Samples 3, 4, 8, 9, and 10).
Overview of gross α determination
The gross α methods included complete destruction of the matrix, evaporation and measurement using α-spectrometry (Sample 7), acid leaching and direct mixing of the resin residue in acid with liquid scintillation cocktail (Sample 3), and acid leaching, filtration, evaporation of aliquot and mixing with liquid scintillation cocktail (Sample 10). Some of these techniques have been published as ISO standards [29,30,31].
Overview of the measurement techniques
Measurement of 241Am, uranium, plutonium, and curium isotopes were carried out using α-spectrometry. Additionally, uranium isotopes in Samples 3 and 8 were also analysed using HR-ICP-MS and ICP-MS/MS, respectively. Gamma spectrometry was also utilised in measurement of 241Am and 235U. The γ-spectrometric analysis results in the previous exercise [4] did not detect these radionuclides most likely due to masking of the low energy signals, such as 241Am (γ-rays with 59.5 keV energy and 35.8% emission probability [32]) by the large number of γ-emitters or due to limit of detection. Nevertheless, γ-spectrometric measurements were carried out for detection of 235U and 241Am in purified fractions (Sample 3) and 241Am in the original and acid digested sample (Sample 10). LSC was utilised in measurement of gross α-activity and 241Pu.
Results and the statistical analyses
The masses taken for the radiochemical analyses varied from 10 to 250 mg (Table 3). Small sample sizes are appropriate in cases when the samples are homogenous and activity concentrations are high enough for quantitative analysis. In this study, the particle size of the sample ranged from 10–200 um. Whether small subsamples of < 100 mg can be considered to be representative is dependent on the homogeneity of the material at the sample size used, and must be considered on a case-by-case basis, since the homogeneity measurements described earlier were carried out with samples of approximately 0.8 g, and are not strictly applicable to smaller sample sizes. Furthermore, the relative uncertainty of mass measurements at low sample masses may become comparable to other contributing uncertainties.
The acid digestions resulted in total destruction of the matrix in all cases except for Samples 2, 6 and 10, which were filtered prior to radiochemical separations. Sample 2 had been treated with aqua regia and H2O2 on a hot plate. Sample 6 had been treated with mixture of H2SO4, HNO3 and HClO4 on a sand bath. Sample 10 had been treated with Fenton’s reagent and after filtration, the residue was treated with HNO3. The complete destruction of the matrix was achieved with HNO3 and H2O2 mixture in a microwave oven (Sample 3), HCl, HNO3 and H2O2 mixture on a hot plate (Sample 4), HNO3 treatment in a microwave oven (Samples 5 and 9), HNO3 and HCl mixture in a microwave oven (Sample 7), and HNO3, HCl, and H2O2 mixture under reflux (Sample 8). The success of complete destruction of the matrix does not seem to be linked with sample masses (Table 3) nor acids whereas a link can be drawn with use of a microwave oven in which temperature, pressure, acid and time contribute to the decomposition process. Complete destruction of the matrix had been also achieved without a microwave oven when the sample had been pre-treated with drying and ashing (Sample 4) and under a reflux (Sample 8). The ashing had been carried out at 500 °C in order to destroy the organic matter through combustion of the styrene–divinylbenzene copolymer structure of the resin prior to the acid digestion. At the above-mentioned temperature, loss of actinides or formation of difficult to dissolve species should not occur. However, complete destruction of the matrix is not necessary when the studied radionuclides are quantitatively extracted using acid leaching. For example, weak acid resins have a high affinity for the H+ and are consequently easily regenerated with strong acids whereas strong acid resins retain their ability to exchange cations across the entire pH range. Use of oxidising acids such as HNO3 attack the resin structures causing degradation of the matrix and consequently both regeneration and degradation of the resin occurs. Therefore, the resin specifications should be considered in selection of the acid treatment method. In this study, the resin was a mixture of strong acid cation and strong base anion exchange resins and consequently complete destruction of the matrix is advisable even though no causation can be seen between the activity concentration results of the samples with complete and incomplete acid digestions. Complete destruction of the matrix could be also carried out using alkali fusion, which is an efficient method for treatment of large sample sizes (e.g., in analysis of low activity samples).
Table 3 summarises the preliminary uranium, plutonium, and americium yield results. The results show that the uranium yields were mainly good varying between approximately 60% to 80% whereas wide range of yields were reported for plutonium (from estimated < 1% to approximately 100%) and americium (from < 1% to 110%). The Sample 6 yield results suffered from significant challenges in their determinations and consequently the yield were estimations. The curium results were calculated from the americium yields, as 243Am was used as a tracer for Cm isotopes as well. Longer lived curium isotopes are not available as tracers for radiochemical analysis of 242,243,244Cm, and therefore 243Am is commonly used as a tracer isotope in analysis of both 241Am and 242,243,244Cm, due to adequately similar chemical behaviour [33] of these two elements.
Uranium isotopes
The final uranium isotope analysis results were not statistically analysed as only four data entries above limit of detection for each radionuclide, 234U, 235U and 238U, were available. However, an interesting phenomenon in the uranium isotope measurement capabilities were seen in the preliminary and final results when comparing γ-, α- and mass spectrometry. Mass spectrometry determinations of uranium isotopes return lower detection limits than radiometric techniques, as demonstrated in Sample 3, where 235U results by γ-and α-spectrometry were below limits of detection, 5.3 Bq g−1 and 40 mBq g−1, respectively, although it was possible to detect 235U quantitatively by mass spectrometry, where the activity concentration was determined to be 8 µBq g−1. However, quantitative 234U and 238U results were achieved both with α-(Sample 5) and mass (Sample 3 and 8) spectrometry at mBq g−1 level.
Plutonium isotopes
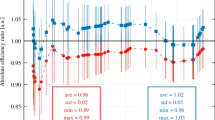
Statistical analysis was carried out for the final 238Pu and 239,240Pu results (Table 4). Sample 6 results were not included in the assigned value calculation as they were reported late. Therefore, the 238Pu and 239,240Pu assigned values, 2.2 ± 0.6 Bq g−1 and 420 ± 150 mBq g−1 respectively, were both calculated from six data entries. The robust standard deviation of 238Pu results was 14% and this value was used in the z-score assessments. The corresponding 239,240Pu result was 34% and consequently standard uncertainty of assigned value in the z-score assessments (Eq. 2). The final 238Pu results with the assigned value in Fig. 1 show a good general trend even though sample 3 has been given a warning signal, where 2.0 <|z|< 3.0, and sample 6 is unacceptable, where |z|≥ 3.0, as seen in Table 5. The final 239,240Pu results in Fig. 2 and Table 5 show that sample 3 and 4 deviate a little from the general trend whereas sample 6 also deviates from the general trend and has very large uncertanties. Sample 3 has been given a warning signal whereas sample 4 and 6 results are in the unacceptable range with |z|≥ 3.0.

Final 238Pu results and assigned value in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2

Final 239,240Pu results and assigned value in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2
Statistical analysis was not carried out for the final 241Pu results as only four data entries were available. The comparison of preliminary and final 241Pu results in Fig. 3, however, show significant improvement in the general trend after the Sample 3 and 4 results had been re-assessed. The preliminary Sample 3 result was an order of magnitude greater than the final result whereas the preliminary Sample 4 results were approximately four times higher than the final result. Possible reasons for the deviations of the preliminary Sample 3 and 4 results were suggested to originate from the LSC efficiency calibrations or spectral interferences. A general practice in 241Pu LSC measurements is to use 3H in LSC efficiency calibrations due to difficulties in procurement of 241Pu standard solutions. Although the β-energy maxima for 241Pu and 3H are similar, it should be noted that this approach only approximates the efficiency calibration for 241Pu, due to significant differences between the shapes of the β-spectra, and hence detection efficiency, for 241Pu and 3H. Additionally, it was suggested that the spectral interferences may rise from 60Co contamination or color quenching due to acid stripping of plutonium measurement sources after α-spectrometric measurements. However, 241Pu measurements of Sample 3 were carried out in aliquots taken prior to electrodeposition and consequently it was proposed that the yield determination in the electrodeposited measurement source using α-spectrometry was not necessarily the same as in the 241Pu aliquot. Therefore, the final 241Pu results of Sample 3 were measured after acid stripping of two plutonium measurement sources and the yields were re-calculated after α-spectrometry measurement of the acid stripped measurement sources. The same LSC 3H measurement protocol was used but an improved feature to limit spillover of α-signal into the β-channel was implemented. Consequently, the 241Pu results for sample 3 were reduced to a comparable level. The improvement in the 241Pu result for sample 4 originated from a re-calculation of the result after discovery of impurities in the 242Pu source. The partner had concluded that their preliminary results were systematically above the general trends and after inspection of their procedures noticed that the 242Pu tracer included 238Pu, 239,240Pu, 241Am and 241Pu impurities. Consequently, the partner re-calculated all their Sample 4 results taking into consideration the effect of the impurities.

Preliminary and final 241Pu results in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2
241 Am
Two types of statistical analyses were carried out for 241Am results, namely statistical analysis according to the ISO 13528 standard and the extended data analysis. First, the statistical analysis was carried out for the final 241Am results according to the ISO 13528 standard. The 241Am assigned value of 1.2 ± 0.4 Bq g−1 was calculated from six data entries (Table 4). Sample 6 result were not included in the assigned value calculation as it was reported late. The robust standard deviation was 18% and therefore it was used in the z-score assessments. The final 241Am results with the assigned value in Fig. 4 show good consistency and consequently all z-score results (except sample 6) are in acceptable range (Table 5).

Final 241Am results and assigned value in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2
Preliminary 241Am results measured using γ-spectrometry were submitted for Samples 3 and 10. In both cases, the results were below limit of detection of 89 Bq g−1 and 5 Bq g−1, respectively. Sample 3 measurements were carried out in the purified americium fraction, which benefit is in the elimination of interfering radionuclides. However, in the Sample 3 measurements, the sample size was small (50 mg) and 54Mn, 134Cs and 137Cs were seen in the γ-spectrum. Additionally the yield calculations were carried out based on the electrodeposited tracer and, as discussed earlier for 241Pu sample 3 measurements, the yield prior and after electrodeposition is not necessarily the same. The limit of detection for the Sample 10 measurements using γ-spectrometry was significantly lower than Sample 3. Sample 10 measurements were carried out in the acid digested solution in which 0.25 g of resin had been partially decomposed. However, the γ-spectrometric 241Am quantification was challenged by low 241Am activity and the high Compton background arising notably from presence of 60Co and 137Cs.
Second, the extended data analysis for the 241Am results were analysed using a combination of ζ-score, z-score and relative uncertainty test as described earlier. Sample 6 result was a clear outlier. However, the dataset was too small to make this approach meaningful although some useful information can be extracted. If the means of the results for 241Am are computed, the results are shown in Table 6.
The result of Sample 2 might be considered an outlier, but rejection of this data point from a dataset of only six results is questionable. However, if this is done, the outcome is shown in Table 7.
The outcome of this analysis is to shift the means slightly, and in the case of the Mandel-Paule mean to expand the uncertainty by forcing the reduced \({\upchi }^{2}\) value to 1 by adding additional uncertainty to each of the contributing values and so the Mandel-Paule mean [34, 35] and uncertainty are reasonable estimates for the assigned value and uncertainty.
Curium isotopes
Statistical analysis was carried out for the final 243,244Cm results (Table 4). The 243,244Cm assigned value of 5.4 ± 2.9 Bq g−1 was calculated from six data entries. Sample 6 result was not included in the assigned value calculation as it was reported late. Both the robust standard deviation and standard uncertainty of assigned value were significantly large –53% and 27%, respectively. However, for the sake of the exercise, the standard uncertainty of assigned value was used in the z-score assessments. The final 243,244Cm results with the assigned value in Fig. 5 show a scattered trend. Consequently, only one z-score result (Sample 2) was in the acceptable range, where |z|≤ 2.0, whereas one result was unacceptable and all the other results were given a warning signal (Table 5). The scatter was postulated to originate from slightly different chemical behaviour of the americium and curium, because even though the same americium tracer was used in the yield calculations, the 241Am results were well comparable whereas 234,244Cm results were not. Several different types of radiochemical methodologies were utilised in this exercise and consequently different chemical behaviour of americium and curium can result in significantly different outcomes. However, the results were in same order of magnitude between 2 and 8 Bq g−1.

Final 243,244Cm results and assigned value in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2
Statistical analysis of the final 242Cm results were not carried out as only four results above limit of detection were available. However, the preliminary 242Cm results (Fig. 6) show inconsistencies similarly to the 243,244Cm results. For example, the highest and lowest 242Cm results were below limit of detection. Critique was given to the reliability of the results due to the short half-life of 242Cm, 162.9 d [36]. It was found in a previous NKS intercomparison exercise, that decay corrections for 242Cm might be problematic, leading to highly varying results among laboratories, not only due to the half-life of 242Cm, but also because of unknown activity content of its parent nuclide 242mAm (half-life 141 a) in a nuclear power plant sample [23, 37]. Additionally, the use of americium tracer in the yield calculations needs to be considered as previously discussed.

Preliminary 242Cm results in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2. Red dot indicates limit of detection result
Gross α activity measurements.
Statistical analysis of the gross α-activity results was not carried out as only three analysis results were available. However, a significant improvement in the gross α-activity results can be seen between the preliminary and final results in Fig. 7. The preliminary results show that Sample 3 gross α-activity is approximately three times higher than the Sample 7 and 10 results. It was suggested the Sample 3 measurement results may have suffered from spillover of the β-signal to the α-side. The presence of much higher levels of β-emitters compared to α-emitters may be present in spent primary circuit ion exchange resins due to the source of the radionuclides in that β-emitting radionuclides is mainly from corrosion of primary circuit steel piping internals and α-emitting radionuclides from spent fuel leakage. Therefore, even low levels of spillover from the β-channel to the α-channel can cause a significant bias. The LSC spectrum of the Sample 3 gross α-activity results were re-studied and a 2D plot (Fig. 8) revealed the erroneous counting of β-coincidence signal as α-particle signal shown as pulses on the upper edge of the spectrum. The coincidence phenomenon was counteracted by lowering quenching with addition of more liquid scintillation cocktail and the Sample 3 was remeasured using a ‘spill to beta’ parameter in the Hidex 300SL counter. The pulse length index (PLI) parameter, which discriminates between α- and β-signals, was kept as default setting and the triple to double coincidence ratio (TDCR) was directly used as measurement efficiency of 100% even though more reliable results would have needed a more exhaustive efficiency correction. However, with these corrective actions, the final Sample 3 results were well aligned with the other data entries.

Preliminary and final gross α-results in spent ion exchange resin with uncertainties stated with a coverage factor of k = 2. Red dot indicates limit of detection result

Preliminary and final 2D plots of gross β-activity measurement of Sample 3. The x-axis is channel and y-axis is pulse length index
An additional study was carried out after the intercomparison exercise to test the significance of removal of the overwhelming number of β-emitters in the gross α-LSC measurement. An α-PSresin is a new plastic scintillation resin which has been reported for an analysis of gross α-activity in drinking and river waters [38]. A set of α-PSresin was kindly provided as pre-packed columns, which were first tested for yield and determination α- and β-separation parameter (PLI) using 233U and 90Sr/90Y standards. The columns were treated in a vacuum box and flowrate was preferably kept below 1 ml per minute. Stable europium (1 mg) was used for the yield determination, which was 100% in all cases. Two α-PSresin columns were treated with known amounts of 233U and 90Sr/90Y standards (90Y retained in the column) to determine the PLI level. After establishment of the PLI level, two 10 mg resin samples were first acid digested with 1 mg Eu using 8 M HNO3 for 2 h at 200 °C. The samples were filtered through Whatman glass microfibre filters and diluted to 6 M HNO3 using deionised water prior to β-emitter removal in pre-treated α-PSresins. The α-PSresin separation consisted of pre-treatment of the pre-packed colums with 6 M HNO3, introduction of the sample, and washing of the columns using 6 M HNO3 and deionised water. Drying of the columns were carried out by reducing the pressure inside the vacuum box for 10 min. The yield determinations were carried out by taking ICP-OES samples from the flow through solution and measurement of Eu. The gross α-activity measurements were carried out by placing the α-PSresin columns inside plastic LSC vials and measured using pre-determined gross α-activity measurement parameters. The initially measured averaged gross α-activity result was 36 Bq g−1, which was approximately three times higher than the previously determined Sample 3 results. The samples were re-measured every few days and a decrease in activity was observed and contributed to ingrowth and decay of 90Y decay. The measurement results stabilised to 9 Bq g−1 after couple of weeks giving comparable results (see Fig. 7).
Discussion
The radiochemical analyses of α-emitting radionuclides are as challenging as analyses of β-emitting radionuclides. In both cases, interfering radionuclides and stable elements must be preferably removed or at least minimised using precipitations, ion exchange and chromatographic column separations. However, many of the α-emitting radionuclides can be present in several oxidation states causing an additional challenge in management of their chemical behaviour. Similar to the detection of the β-emitters, α-emitter measurement sources require careful preparation based on the detection technique. The following sub-sections present critical considerations in the measurement source preparation, analysis of americium and uranium, plutonium, curium isotopes, gross α-activity analysis, and intercomparison exercises and statistical analysis.
Critical considerations in the measurement source preparation
Alpha particles are relatively large and strongly ionising particles with a short travel range in material and air. Therefore, they are prone to self-absorption, which means that their α-decay energy is absorbed by collisions with surrounding material. In the case of α-spectrometric measurements, the α-particle self-absorption hinders the signal travel from the sample surface to the detector. Therefore, the most important part in preparing a counting source for α-spectrometric measurement is to have a smooth and as thin as possible, almost massless, measurement source. Otherwise, high resolution, derived from the full width at half maximum (FWHM), spectrum peaks cannot be obtained. The resolution is very sensitive to the source mass, for example increasing from 40–50 keV to around 70 keV, when the solid deposition is increased from < 100 µg to 100–200 µg [39]. Sample purity is important as well, since any impurities present in the sample will increase the source thickness, causing tailing and degradation of the resolution.
Before the measurement source preparation, it is essential to separate the analyte of interest from other radionuclides. Otherwise, the energy peaks of the different radionuclides will overlap, and the activity of a certain radionuclide cannot be reliably determined. Another thing to consider in the source preparation is the deposition yield—the fraction of the tudied analyte that is deposited from the solution onto the measurement source [40] Deposition yields vary between different source preparation methods in that one method can be more suitable for some analytes but give poor yields for another [27, 40].
The most common methods for α-spectrometry source preparation are micro-coprecipitation and electrodeposition, both used in this intercomparison project. For micro-coprecipitation, a lanthanide carrier is added to the actinide solution, and both the radionuclide of interest and the carrier are precipitated as fluorides with addition of concentrated HF [41]. Actinides with higher oxidation states, including uranium and plutonium, require the use of a reducing agent, such as TiCl3, for a more complete precipitation. The precipitate is left to form at a low temperature for at least 20 min [42] before filtration on to a membrane or micro-pore filter paper. During the filtration, a substrate solution of the lanthanide is filtered first to form a base layer for the actinide precipitation. This produces a more even deposition and prevents the actinides entering the pores of the filter, resulting in a higher resolution [42]. If filtration is performed under a vacuum, instead of a syringe filtration, the actinides are likely to be retained deeper in the pores of the filter, causing peak tailing in the spectrum [43]. Care should be taken in the removal of impurities, such as iron and carbon, since micro-coprecipitation is not a specific method, and any impurities present can precipitate on the source as well [44, 45].
In electrodeposition, the analyte deposited from solution on a stainless-steel disk with the use of an electrical current. The method is more time-consuming than micro-coprecipitation, one sample taking around 1–3 h [20, 27]. The dry actinide samples are usually dissolved in a H2SO4 solution, and the solution is transferred into the electrodeposition cell [27]. No HCl should be present in the deposition cell, as it can corrode the electrodes and thus affect the quality of the deposition [20]. The pH of the system needs to be carefully adjusted and maintained during the electrodeposition, typical pH being around 2–3 [27]. The discs are rinsed after the deposition and dried. The method is very sensitive for changes in the electric current and the pH, as well as any impurities present in the sample solution. The impurities, such as metal ions and carbon, are competing for deposition sites with the actinides and can lead to lower deposition yields or peak tailing [20, 44, 46]. Adjustment of the pH and keeping it steady can be time-consuming, and it requires skill [27, 40]. An NaHSO4-Na2SO4 solution has been used as an electrolyte since it is self-buffering at pH ~ 2 and remains stable during the deposition [20]. Sodium hydrogen sulphate is also useful in evaporation steps prior to electrodeposition because it prevents actinides from adsorbing on beaker walls, increasing the radiochemical yield [20].
Electrodeposition usually gives better resolution than micro-coprecipitation, however, the latter still has sufficient resolution to distinguish between 244Cm and 241Am peaks in the same spectrum [43]. In addition, electrodeposition has a lower reproducibility for high deposition yields [43]. Micro-coprecipitation may be a more practical option if a large number of samples are measured [44].
In this study, the sample preparation was discussed but no discussions were carried out for visual inspections of spectra. Therefore, no assessments on the success of the sample preparations using FWHM were carried out.
Critical considerations in the uranium isotope analysis
Uranium-234, 235U and 238U can be quantified by α-spectrometry or ICP-MS. Alpha spectrometry has been successfully utilised in determination of uranium isotopes from environmental and mining samples [25, 47, 48] and nuclear power plant and nuclear reactor waste samples [26, 49,50,51] during the past decades. As it was mentioned in the previous section, a thin α source is essential in determination of uranium isotopes by α-spectrometry. Besides separating uranium efficiently from stable elements which are often abundant in decommissioning and environmental samples in much higher concentrations than actinides, it is also necessary to separate actinide elements efficiently from each other, due to their similar α-energies [49,50,51,52]. An adequately pure uranium sample for α-spectrometric purposes may be achieved by ion exchange, extraction chromatography and precipitation, and usually a combination of these techniques is employed.
Uranium has two main oxidation states, U(IV) and U(VI), in environment. In oxygen-rich environment and in acidic solutions, uranium is predominantly in U(VI) form. However, during radiochemical separations and as already mentioned in α source preparation by lanthanide fluoride coprecipitation, U(VI) can be converted to U(IV) by adding a suitable reductant, such as TiCl3, ascorbic acid, and Fe(II) compounds to the sample solution. Usually, the oxidation state of reduced uranium returns quickly to an oxidised form, being U(VI) in radiochemical separations. Despite the advantages of α-spectrometry, ICP-MS can be considered superior for analysis of long-lived uranium isotopes, although one should note that elemental purity requirements are quite different for both measurement techniques. ICP-MS enables to achieve a better sensitivity in a shorter counting time in comparison to α-spectrometry, and is used widely for the routine analysis of uranium [1]. As for α-spectrometry, a radiochemical purification must be applied before ICP-MS measurement, especially to remove plutonium for accurate 238U analysis, when excessive quantities of 238Pu is present. Otherwise, presence of isotope 238Pu in the sample causes isobaric interference on 238U in mass spectrum. For that purpose, the purification step is nowadays often based on extraction chromatography with UTEVA® resin [28, 28, 50, 54] since it separates efficiently uranium from other actinides with lower oxidation states. Also, the column separation with UTEVA® resin is relatively fast compared to ion exchange. However, other extraction chromatography resins as well as ion exchange are also used for separating uranium for mass spectrometric measurements. Recently, new developments to simplify the radiochemical procedures have been carried out by reducing isobaric interferences with the implementation of collision/reaction cells available in ICP-MS devices [53] or the use of ICP-MS/MS instruments [28, 54].
Regardless of the detection method, the radiochemical separation methods are still needed to achieve unbiased results for uranium analysis. In the present intercomparison exercise, the majority of the laboratories used 232U as tracer and UTEVA® and/or TRU® resins to purify a uranium fraction. All laboratories (except Sample 6) obtained separation yields higher than 50% which demonstrated their competence in implementing the radiochemistry method dedicated to uranium purification. The measurements of uranium isotopes were mainly performed by α-spectrometry. Sample 3 and Sample 8 were analysed in the purified uranium fractions respectively by HR-ICP-MS and ICP-MS/MS and obtained better detection capabilities in comparison to α-spectrometry, which was very interesting in characterisation of all uranium isotopes in the studied resin sample.
Critical considerations in Pu isotopes analysis
All plutonium isotopes are radioactive, and for environmental and nuclear waste samples, the main nuclides of interest are 238Pu, 239Pu, 240Pu and 241Pu [1, 52]. Plutonium isotopes have been characterised in various types of radioactive waste, such as resins [50, 55,56,57,58], charcoal beds [50], metals [59], evaporator concentrates [55, 57, 60] and effluents [58, 61], noting that 241Pu is a β-emitter while other plutonium isotopes are α-emitters. For all plutonium isotopes, a radiochemical purification procedure must be applied before measurement. Coprecipitation, liquid–liquid extraction, ion exchange chromatography and extraction chromatography are often used for plutonium purification, although liquid–liquid extraction is rarely used in analysis[59]. Anion exchange resin has been particularly applied in HCl or HNO3 medium [55,56,57,58,59, 62]. Different extraction chromatographic resins can be implemented, such as TRU® [55, 61], TEVA® [61] and DGA® [60] resins. The radiochemical procedure used for α-emitting isotopes of plutonium determination can be implemented for 241Pu analysis but adaptations can be carried out depending on the detection technique (for example, LSC or ICP-MS). In the present work, only two laboratories (Sample 2 and Sample 7) performed a coprecipitation step based, respectively, on iron hydroxide in NaOH medium and lanthanum hydroxide in ammonia medium to isolate plutonium isotopes. Sample 2 was prepared with a micro coprecipitation with neodymium trifluoride to prepare an appropriate source for α-spectrometry. Anion exchange chromatography was implemented for Sample 7, Sample 9 and Sample 10. Most of the laboratories carried out separations based on extraction chromatography and combined different types of resins (TRU®, TEVA® and DGA®) to achieve high decontamination factors.
Furthermore, the activity concentrations of natural α-emitting radionuclides (uranium, thorium, polonium) are often orders of magnitude higher compared to the activity concentration of artificial α-emitters especially in environmental samples. Therefore, it is crucial to reduce the concentration of natural α-emitters to a tolerable level prior to determining plutonium isotopes by α-spectrometry. It is even more important in ICP-MS determination of plutonium isotopes, because 238U and its hydrides are isobaric interferences in determination of 239Pu, 240Pu and especially 238Pu with ICP-MS.
It must be noted that plutonium chemistry is complex due to the existence of various oxidation states, which complicates the overall separation process [63]. Plutonium can be encountered in four oxidation states (III, IV, V, VI) in solution, which affects its behaviour during the different purification steps. For example, Pu(III) is not retained on anion exchange resin in HCl medium whereas Pu(IV) and Pu(VI) are strongly fixed in the same conditions. Pu(IV) can polymerise in nitric acid depending on plutonium and nitric concentrations. Consequently, the main challenge is the adjustment of oxidation state during the radiochemical separation method. Success or lack of it affects the overall radiochemical yields and thus the sensitivity. Different chemical reagents have been applied in the literature. The main reagent used for plutonium oxidation is sodium nitrite [55, 56, 58, 59, 61, 64]. Plutonium reduction is often performed with sodium sulphite, hydroxylamine hydrochloride [50, 55], oxalic acid [61], ascorbic acid [50, 65] and hydroiodic acid [55, 56, 62]. The completion of the redox cycle is a key step in successful radiochemistry for plutonium. A failure of this process can induce low separation yields, as it might be the case for Sample 5 and Sample 6. In the present work, Sample 2, Sample 3 and Sample 7 used sodium nitrite and hydroxylamine hydrochloride for plutonium redox cycle.
In this intercomparison exercise, 239Pu and 242Pu tracers were used to determine plutonium separation yields. As mentioned in the previous sections, special care has to be taken towards the choice of tracers to avoid errors in determining the purification yield. Sample 4 highlighted that the tracer purity is essential to garantee accurate results. Actually Sample 4 observed 238Pu, 239,240Pu, 241Am and 241Pu impurities in 242Pu tracer which induced unacceptable results when no correction was applied to take into account of the impurities.
As α-emitters, 238Pu, 239Pu and 240Pu can be determined by α-spectrometry. The same critical considerations as for uranium analysis by α-spectrometry can be applied towards the preparation of thin sources for plutonium isotopes and other α-emitting radionuclides (uranium, thorium, polonium). It must be noted that α-decay energies of 239Pu (5106 keV, 11.9%; 5144 keV, 17.1%; 5157 keV, 70,8%) [66] and 240Pu (5124 keV, 27.2%; 5168 keV, 72.7%) [32] are so close that they cannot be resolved from each other in routine α-spectrometry, and so usually only the sum activity of 239Pu and 240Pu can be measured. However, from a thin electrodeposited source and by using spectral deconvolution programs, it is possible to resolve the α-peaks of 239Pu and 240Pu from each other [52], although this technique might not be suitable for routine analysis. A more common method for determining separate concentrations of 239Pu and 240Pu is to measure them by ICP-MS. This technique does not only enable separate detection of 239Pu and 240Pu and their mass ratio 240Pu/239Pu, but also it has a much lower detection limit for long-lived plutonium isotopes and much shorter measurement time, compared to α-spectrometry. However, 238U and its hydrides can generate isobaric interferences towards 239Pu and 240Pu. The presence of 238U highly hinders 238Pu quantification by ICP-MS through direct isobaric interferences. Therefore, α- and mass spectrometry can be considered complementary in analysis of plutonium isotopes [1]. For both techniques, a highly selective purification method is required before plutonium measurement. As for uranium analysis, the application of collision/reaction cells available in ICP-MS instruments [67] or ICP-MS/MS [86] systems can be considered to simplify the purification methods, but those approaches are still not widespread for plutonium analysis in nuclear waste. In the present work, only α-spectrometry was used for determination of α-emitting isotopes of plutonium. It can be noticed that despite the variety of purification methods, the results related to plutonium α-emitters are relatively consistent (robust standard deviation percentages of 14% for 238Pu and 34% for 239,240Pu), which highlighted the validity of the results obtained by the laboratories that worked on α plutonium characterisation. Radioanalytical problems in plutonium separations due to natural radionuclides and oxidation state adjustment has been also discussed in connection with a previous NKS intercomparison exercise [44].
Plutonium-241 is an isotope of plutonium formed when 240Pu captures a neutron. It is a β-emitter (half-life 14.33 years, βmax 20.8 keV) [66] decaying to 241Am. This radionuclide has a short half-life contrary to 241Am (432.6 years) [32]. Plutonium-241 can be indirectly determined by measuring its decay product 241Am by γ-spectrometry. For its direct measurement, 241Pu must be isolated from the matrix and all the other interfering radionuclides, in particular β-emitters, before LSC measurement. As 241Pu is a low β-emitter, it is possible to use 3H to establish the efficiency curve for 241Pu quantification [68] to avoid the supply of 241Pu sources. In the RESINA project, the separated plutonium used for determination of 238Pu and 239,240Pu is also analysed by LSC for 241Pu measurement after the α-spectrometric analysis. The present intercomparison exercise demonstrated that 241Pu analysis is more difficult than 238Pu and 239,240Pu quantification. During the preliminary meeting, the four results submitted initially were scattered and two laboratories (Sample 3 and 4) obtained much higher values. The latter two conducted investigations to understand where the difference in results came from. As mentioned above, Sample 4 had been analysed with a tracer containing impurities at significant level which introduced a bias. Sample 3 had been initially analysed by taking an aliquot prior to electrodeposition. Sample 3 was re-analysed using acid stripping of the plutonium measurement source and re-measurement of the acid stripped measurement source for yield corrections. Additionally, the LSC measurement parameters using 3H efficiency correction was modified to accommodate interference from α decay. After those investigations, the four results were found to be in the same order of magnitude, which is reassuring for future 241Pu analyses.
Critical considerations in the americium and curium isotope analysis
Americium and curium isotopes in the spent nuclear fuel originate from the neutron activation of uranium or plutonium isotopes. The main americium and curium isotopes in spent fuel and found in spent ion exchange resins are 241Am (432.6 a) [32], 242Cm (162.9 d) [36], 243Cm (28.9 a) [36] and 244Cm (18.1 a) [36]. Americium-241 is the main contributor of decay heat in spent fuel after the first 100 year of cooling [11]. Curium-242 and 244Cm comprise of 90% of all curium isotopes in spent fuel and they have significant contribution to the radiotoxicity [69].
Alpha particle energies of americium and curium isotopes are mostly separated enough for simultaneous measurement with the α-spectrometry. However, 243Cm and 244Cm α-particle energies overlap within about 80 keV energy interval, therefore it is impossible to separate them in conventional α-particle spectrometry. Due to the higher combined activation and fission cross section for 243Cm, the most (< 90%) of the 243,244Cm activity is in fact produced by 244Cm in high burn-up fuels. In addition, 243Cm has also low probability (12.5%) contribution to 242Cm emission at 5992–6067 keV. Therefore, completely accurate quantification of 242Cm would require known concentration of 243Cm. [70].
Americium-243 is often assumed to be a suitable yield tracer for curium isotopes and therefore used as radiochemical yield tracer for their analysis. Both americium and curium are chemically similar and are comparable in ionic size. They both have strong thermodynamic preference to trivalent oxidations state and they are in trivalent state in aquoeus solutions. Habibi et al. [69] conducted speciation computations of different separation steps of americium and curium separation procedure and found out that pH of the solution has significant effect on their speciation during the separation. Even small variations in pH (± 0.5) may result in conditions where americium and curium do not form similar precipitates or complexes, which may induce loss of radionuclide of interest and further biased yields. Hence, pH adjustments during separation and purification procedures should be considered as critical points.
With increasing atomic number in the actinoid series, higher charge of nucleus attracts more 5f electrons and therefore they seldom participate in the bonding processes. Therefore, chemical behaviour of heavier actinides, such as americium and curium, and lanthanides is similar and their prevailing oxidation state is + III. Presence of lanthanides in the sample hinders especially the electrodeposition of americium and curium, leading to a thick α-spectrometry source and further self-absoption and poor quality of measurement source. Problems with deposition quality has been reported previously with americium and curium fractions in intercomparison exercise with samples of nuclear power plant origin [44]. Even though no discussions on the α-spectra were carried out in RESINA project, in some cases of both black and white residues on the metal discs were encountered resulting in poor resolution by tailing to lower energies.
Many of the participants used TRU® extraction chromatography resin for americium and curium and plutonium separation in this exercise. Tri-, tetra- and hexavalent actinides and lanthanides are retained strongly on TRU® in HNO3 media with concentration higher than 0.5 M [71]. Slightly different behavior of americium and curium in the TRU® resin has been demonstrated previously [72]. The heavier neighboring element, curium, has been reported to have lower retention factor to TRU® compared to the lighter neighbor, americium, and a similar phenomenon has been observed in lanthanoid series between Eu(III) and Gd(III) as well [69]. Salminen-Paatero et al. also detected below reference value result for 244Cm, when in the same sample the 241Am results where more evenly distributed below and above the reference value [44]. The use of 243Am as yield tracer can overestimate the curium yields due to the phenomenon where americium retention to TRU® resin is higher than the retention of curium [55, 73, 74].
Also, DGA® resin was used for the americium and curium separation by two of the participants in this exercise. Separation with DGA® resin is less sensitive for Fe(II), Fe(III) competition and retention coefficients greater by factor ≥ 10 compared to TRU® has been reported for americium and curium [75]. However, small differences in the retention behavior have been identified in DGA® resin as well. In nitric acid media the major differences in retention to DGA® are at lower HNO3 concentration ranges (0.01 to 0.1 M) [72] and the use of DGA® resin in higher nitric acid concentration ranges (< 1 M) could result in more uniform americium and curium results compared to TRU®.
Yields for the americium and curium fraction varied significantly (from < 1% to 110%). A poor yield may derive from the competition of Fe(III) and trivalent lanthanides in the TRU® separation step. The yields for DGA® separated americium and curium fraction were both in acceptable range, which could result from the lower sensitivity to competition with Fe(III). Plutonium isotopes may cause spectral interferences, if they are carried over in the americium and curium fraction due to the incomplete oxidation of Pu(III) in the TRU® column. Traces of plutonium in americium and curium spectrum will broaden the α-peaks and will lead to inaccurate results. Habibi et al. [69] tested the use of 248Cm as a yield tracer for curium isotopes successfully. However, the use of 248Cm as a yield tracer may be difficult due to the poor commercial availability.
Critical considerations in the gross α-activity analysis
Even though gross α- and β-activity measurements can be used as a fast screening tool in environment monitoring, radioecology and industrial applications, it has limitations such as loss of volatile radionuclides (e.g., 3H, 14C, radon, 210Po, 137Cs) in cases when heat is included in the sample preparation, changes in sample composition such as total dissolved solids and chemical composition, affect the counting efficiency, and establishment of representative α- and β-particle discrimination where differences between standard solutions and radionuclides in the sample may affect this parameter [76, 77]. Additionally, gross α- and β-activity measurement cannot be used as an absolute determination of the radioactive content in a sample [78]. However, coupled with detailed knowledge of the specific radionuclides present in the sample and their fractional relationship to each other, gross α- and β-activity measurement can be used to calculate the specific content of a radionuclide in a sample of same origin. Gross α- and β-activity analysis is, on the other hand, a quite quick and reliable way to determine the gross level of activity in a sample.
Gas proportional counting (GPC) and LSC are commonly used methods in gross α- and β-activity analysis. The GPC method consists of evaporation of the liquid on a planchet and measurement whereas the LSC method consists of either an evaporation of the liquid into a smaller volume and mixing it with a liquid scintillation cocktail or a direct mixing of a liquid aliquot with the cocktail. Some of the above-mentioned limitations have been counteracted in GPC and LSC analyses for example by preparation of a thin measurement source using a source spraying method [79] and pre-treatment and concentration of the sample solution without heating and removal of β-emitters [38].
Measurement of the gross α-activity with LSC has advantages over specific α nuclide counting [80] as there is a little to no self-absorption in the liquid sample, there are less demands for pre-treatment, sample preparation is less complex, and it is less time consuming. However, the challenges in gross α-activity measurement using LSC are chemical and physical quench, the presence of β-emitters and other interfering nuclides and chemical and physical behaviour of the sample [78, 80]. Quench agents, either chemical or physical, in the sample decrease the counting efficiency and shift the α-spectrum towards the left [81]. The shift in the spectrum due to quench, has the most impact if the sample simultaneously contains both α- and β-emitters. While quenching also leads to a shift in the β-spectrum, the shift in the α-spectra is more pronounced. With both α- and β-emitters present, the spill over of α-signal into the β-spectrum, or vice versa, will cause misclassification and consequently bias in the analysis results.
Other parameters affecting the LSC measurements are for example type of vial and type of scintillation cocktail [77]. The detection efficiency of α-particles in LSC is approximately 100% since α-particles have a short range and display a line spectrum. The pulse height resulting from α decay is a peak at 1/10 of the α particle’s actual energy [82]. The detection efficiency of β decay is less than 100% due to β decay resulting in a continuous spectrum from zero to a maximum energy [83].
The intensity of photons from a scintillation event as a function of time, can be divided into a fast or a prompt component and in a slow or a delayed component. Alpha particles cause a slow delayed component with a decay time of 200–300 ns, while a β-particle causes a fast and prompt component with decay time of a few nanoseconds [83]. The relative amounts of light in the prompt and delayed components are dependent on the specific ionisation of the scintillator and thus is specific to the particle that caused the ionisation. Consequently, differentiation between α- and β-emission by LSC can be carried out and some scintillation cocktails have been designed for enhanced α- and β-particle discrimination. The differentiation between α- and β- pulses is carried out for example using a pulse shape analyser (PSA) or pulse length index (PLI) parameters. However, care should be taken to use the optimal parameter settings, as the analysis is affected by the activity concentrations in a sample, the α- and β- energies and both chemical and color quench level [81, 84]. The optimal parameter setting enables a sufficient discrimination of the α- and β- spectrum and the background effects are reduced. When simultanous measurement of both gross α-activity and gross β-activity in a sample is carried out, care must be taken in the calibration process of the detector to ensure good enough separation of β-signals and α-signals [85]. Additionally, samples containing amounts of higher energy β-particles, can cause spill over into the α-spectrum [81] and thus overestimating the α-activity. The gross α-activity analysis can be carried out without a pulse shape analyser, but more emphasis is needed on elimination of β-emissions [82]. However, real samples may contain several α- and β-emitters with different energies than utilised in the calibrations and different quench properties. Consequently, the optimal parameter settings may not be as precise as desired.
Three of the participants in the RESINA intercomparison exercise performed the gross α-activity measurement. The sample 7 analysis using evaporation and α-spectometry did not suffer from analytical difficulties whereas sample 10 result was comparable with the other results even though it was reported with a limit of detection. The sample 3 analysis initially suffered from significant overestimation of the gross α-activity measurement due to missclassification of overwhelming amount of β-emitters as α-emitters. The missclassification was corrected by lowering quenching with addition of more scintillation cocktail and utilisation of the 'spill to beta' measurement parameter. These two simple modifications enabled a comparable result. Consequently, gross α-activity measurements carried out using LSC (samples 3 and 10) and α-spectrometry (sample 7) produced comparable results. The additional study, in which the α-PSresin was utilised in the removal of overwhelming amount of β-emitters, showed initially a higher gross α-activity due to retained 90Y. However, the result became comparable after decay of 90Y. It can be concluded that some knowledge on which radionuclides may be present in a sample and on what activity level is of importance as the operator can be aware of possible spill overs. Additionally, as increasing quench increases the chances of an α-pulse to be misclassified as a β-pulse, the quench level should be monitored.
Critical considerations in the intercomparison exercises and statistical analysis
The data has been analysed in accordance with the requirements of ISO 13528, with an extended data analysis also used for analysing the data arising from 241Am measurements.
Central to the assessment of the reported data is the selection of a reference or assigned value for each nuclide, since the activity concentration was unknown before these samples were analysed. This may be mitigated by the use of robust values for activity concentrations and uncertainties, but (as an example) the assigned value for 241Am shifts by at least 5% if a suspected outlier is removed. Nevertheless, the assigned values allow an assessment of the data reported.
Determination of assigned values is possible for uranium isotopes, plutonium isotopes (including 241Pu), 241Am and 242Cm. However, knowledge of the ratio between 241Pu and 241Am at a given time better informs the variation of the 241Am activity concentration over time, which will be subject to radioactive decay of 241Am, as well as ingrowth of 241Am from 241Pu decay, although this may not be significant over the duration of this work.
The analysis of gross α-activity and 243/244Cm is complicated by the differing decay rates of the constituent radionuclides, although an approximation can be made for 243/244Cm.
In short, this exercise has provided useful information on the determination of actinide measurement in a challenging matrix, and the outcomes of the data analyses provide good insight if the complexities of such analyses. It would be beneficial to organise a similar comparison exercise involving a wider participation base to allow a more detailed analysis of the measurement data. Additionally, given the importance of the measurements of actinides in radioactive waste, there may be some scope in proposing an extended exercise as a supplementary Bureau international des poids et mesures (BIPM) comparison.
Summary and conclusions
The α-emitting DTM nuclide intercomparison exercise in spent ion exchange resin was carried out using the same methodology as in the previous exercises [2,3,4,5]. The radiochemical procedures in separation and purification of α-emitters consisted of similar techniques as for β-emitters, namely precipitations and different ion exchange and chromatographic resins. However, as majority of the measurements were carried out using α-spectrometry, the preparation of a thin measurement source was of a great importance. Additionally, several α-emitters studied in this exercise can be present in several oxidation states and their control can be concluded to be crucial as they affect both the separation and purification behaviour and measurement source preparation.
The analyses of uranium isotopes were carried out using α- and mass spectrometry. The purification procedures to eliminate interferences were needed prior to both α- and mass spectrometry measurements. In majority of cases good (higher than 50%) yields were obtained. Statistical analyses of uranium isotope results were not carried out as only four data entries above limit of detection were available. However, the results showed that the sensitivity of mass spectrometry is superior to α-spectrometry in uranium isotope detection.
The results related to plutonium α-emitters are relatively consistent, which demonstrates the analytical capabilities of the laboratories towards plutonium analysis by α-spectrometry after radiochemical purification. However, the separation yields need improvement by some laboratories. A poorly controlled redox cycle process applied to control plutonium oxidation state might be the cause of low plutonium recovery yields. Statistical analyses were carried out for the 238Pu and 239,240Pu results, which assigned values were 2.2 ± 0.6 Bq g−1 and 420 ± 150 mBq g−1, respectively. The z-score determinations showed that majority of the reported results were acceptable.
This intercomparison exercise also showed that 241Pu analysis using LSC is much more complicated to manage and is still challenging for some laboratories in comparison to the characterisation of plutonium α-emitters. In particular, special care must be taken towards the establishment of efficiency calibration in LSC, notably when 3H is used instead of 241Pu. The 241Pu counting window has also to be optimised to limit the impact of α-signal and avoid overestimation of 241Pu activity. Moreover, the present intercomparison exercise showed that the purity of the plutonium tracer used must be checked carefully to avoid measurement bias. From those results, significant progress was achieved for 241Pu determination in nuclear waste, which is reassuring for future analysis.
The 241Am results were analysed using statistical analysis according to the ISO 13528 standard and the extended data analysis. The assigned value of 241Am was 1.2 ± 0.4 Bq g−1 and all results (except one) were in acceptable range. The radiochemical purifications of 241Am and curium isotopes are carried out simultaneously and the 234Am tracer is often also allocated for curium yield calculations. However, the results obtained in this exercise clearly demonstrated that use of 243Am tracer is not optimal, due to the slightly different chemical behavior of americium and curium, for curium isotopes since the 243,244Cm results showed large scattering (assigned value 5.4 ± 2.9 Bq g−1) whereas 241Am results were well aligned.
The extended 241Am analysis revealed some interesting information, in that one result was identified as an outlier by the Peirce criterion [12, 13]. However, given the small size of the data set, it is questionable whether outlier rejection can be justified. Use of the Mandel-Paule mean [34, 35] allowed an improved estimate of the assigned value to be made as this mean is weighted by the reported uncertainties with additional uncertainty added to the final value to force the reduced chi squared value to 1. Analysis of the reported uncertainties via the R-test [14] revealed no additional information about uncertainty estimation by the participants. A larger data set would allow more meaningful analysis.
The gross α-activity analyses were carried out using acid digestion/leaching and measurement of evaporated sample on planchet using α-spectrometry or mixing with the leachate or concentrated leachate with liquid scintillation cocktail and measurement using LSC. The results highlighted the significance of careful setting of LSC measurement parameters as the overwhelming quantity of β-emitters in a spent ion exchange resin was causing misclassification of β-signal as α-signal. The additional study with the α-PSresin showed comparable results after removal of β-emitters and decay of 90Y.
As a conclusion, the RESINA intercomparison exercise introduced a new set of challenges in the DTM analyses. Firstly, many of the α-emitters and their interferences can be present in several oxidation states and their control is directly linked with success of the radiochemical purifications and measurement source preparations. Secondly, α and β discrimination settings in LSC measurements are crucial in minimisation of α and β signal misclassifications. Thirdly, radiochemical behaviours of americium and curium isotopes are not necessarily identical and consequently use of 243Am tracer in 243,244Cm analysis may be problematic.
References
Hou X, Roos P (2008) Critical comparison of radiometric and mass spectrometric methods for the determination of radionuclides in environmental, biological and nuclear waste samples. Anal Chim Acta 608:105–139. https://doi.org/10.1016/j.aca.2007.12.012
Leskinen A, Tanhua-Tyrkkö M, Kekki T, Salminen-Paatero S, Zhang W, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Stordal S, Isdahl I, Fichet P, Gautier C, Brennetot R, Lambrot G, Laporte E (2020) Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-429, Roskilde, Denmark. ISBN 978–87–7893–519–9. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214696476
Leskinen A, Tanhua-Tyrkkö M, Salminen-Paatero S, Laurila J, Kurhela K, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Laporte E, Guiliani M, Bubendorff J, Fichet P (2021) DTM-Decom II - Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-441, Roskilde, Denmark. ISBN 978–87–7893–533–5. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214697245
Leskinen A, Dorval E, Salminen-Paatero S, Hou X, Jerome S, Jensen K-A, Skipperud L, Vasara L, Rautio S, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Baudat, E., Lambrot G, Giuliani M, Colin C, Laporte E, Bubenforff J, Brennetot R, Wu S-S, Ku H, Wei WC, Li YC, Luo QT (2022). DTM-Decom III - Intercomparison exercise in analysis of DTM beta and gamma emitters in spent ion exchange resin. NKS-457, Roskilde, Denmark. ISBN 978-87-7893-550-2. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214697836
Leskinen A, Lavonen T, Dorval E, Salminen-Paatero S, Meriläinen V, Hou X, Jerome S, Jensen K-A, Skipperud L, Rawcliffe J, Bourgeaux-Goget M, Wendel C, Stordal S, Isdahl I, Gautier C, Taing Y, Colin C, Bubendorff J, Wu S-S, Ku YH, Li YC, Luo QT (2023) RESINA – Intercomparison exercise on alpha radionuclide analysis in spent ion exchange resin. NKS-466, Rosklide, Denmark. ISBN 978–87–7893–560–1. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214698260
Leskinen A, Salminen-Paatero S, Gautier C, Räty A, Tanhua-Tyrkkö M, Fichet P, Kekki T, Zhang W, Bubendorff J, Laporte E, Lambrot G, Brennetot R (2020) Intercomparison exercise on difficult to measure radionuclides in activated steel: statistical analysis of radioanalytical results and activation calculations. J Radioanal Nucl Chem 324(3):1303–1316. https://doi.org/10.1007/s10967-020-07181-x
Leskinen A, Gautier C, Räty A, Kekki T, Laporte E, Giuliani M, Bubendorff J, Laurila J, Kurhela K, Fichet P, Salminen-Paatero S (2021) Intercomparison exercise on difficult to measure radionuclides in activated concrete - statistical analysis and comparison with activation calculations. J Radioanal Nucl Chem 329(2):945–958. https://doi.org/10.1007/s10967-021-07824-7
Leskinen A, Dorval E, Baudat E, Gautier C, Stordal S, Salminen-Paatero S (2023) Intercomparison exercise on difficult to measure radionuclides in spent ion exchange resin. J Radioanal Nucl Chem 332(1):77–94. https://doi.org/10.1007/s10967-022-08687-2
International Standard ISO 13528:2022, Statistical methods for use in proficiency testing by interlaboratory comparison. ISO, Geneva, Switzerland
Committee on Radiation Protection and Public Health. (2014). Radiation protection aspects of primary water chemistry and source-term management. OECD report NEA/CRPPH/R(2014)2. https://one.oecd.org/document/NEA/CRPPH/R(2014)2/En/pdf
Feiveson H, Mian Z, Ramana MV, von Hippel F (eds) (2011). Managing spent fuel from nuclear power reactors: experience and lessons from around the world. International Panel on Fissile Materials report. https://fissilematerials.org/library/rr10.pdf
Peirce B (1852) Criterion for the rejection of doubtful observations. Astron J 2(21):161–163. https://doi.org/10.1086/100259
Ross SM (2003) Criterion for the rejection of doubtful observations. J Eng Technol 20(2):38–41
Jerome SM, Harms AV (2023) Proficiency test data interpretation and data rejection. Appl Radiat Isot 194:110678. https://doi.org/10.1016/j.apradiso.2023.110678
Calin MR, Saizu MA, Radulescu I (2013) Assets on energy and efficiency calibration of an alpha spectrometry system using standard sources. J Radioanal Nucl Chem 298(1):55–66. https://doi.org/10.1007/s10967-013-2474-2
Chen QJ, Aarkrog A, Nielsen SP, Dahlgaard H, Nies H, Yu Y, Mandrup, K (1991) Determination of plutonium in environmental samples by controlled valence in anion exchange. Risø National Laboratory Report Risø-M-2856. https://backend.orbit.dtu.dk/ws/portalfiles/portal/7783868/RISOM2856.pdf
Eichrom Technologies (2014) Americium in water, Analytical Procedure, Method No ACW04. https://www.eichrom.com/wp-content/uploads/2018/02/acw04-13_am-water.pdf
Hou X, Anderot M, Ekerljung L, Salminen-Paatero S, Olszewski G, Öhlin H, Davidsson M, Bruzell F, Tovedal A, Rautio S, Allard S, Hatakka A, Wijk S, Eriksson M, Åberg H, Settervik E, Suutari T, Vesterlund A, Lindahl P, Kangas S, Hovi P (2020) Determination of important alpha emitters in nuclear and environmental samples—OptiMethod 2019 project report. NKS-436, Roskilde, Denmark. ISBN 978-87-7893-526-7. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214696641
Ikäheimonen TK (2000) Measurement of 241Pu in environmental samples. J Radioanal Nucl Chem 243(2):535–541. https://doi.org/10.1023/A:1016023524695
Kressin IK (1977) Electrodeposition of plutonium and americium for high resolution α-spectrometry. Anal Chem 49(6):842–846. https://doi.org/10.1021/ac50014a044
López-Lora M, Levy I, Chamizo C (2019) Simple and fast method for the analysis of 236U, 237Np, 239Pu and 240Pu from seawater samples by accelerator mass spectrometry. Talanta 200:22–30. https://doi.org/10.1016/j.talanta.2019.03.036
López-Lora M, Chamizo E, Villa-Alfagem M, Hurtado-Bermúdez S, Casacubert N, García-León M (2018) Isolation of 236U and 239,240Pu from seawater samples and its determination by accelerator mass spectrometry. Talanta 178:202–210. https://doi.org/10.1016/j.talanta.2017.09.026
Salminen-Paatero S, Meriläinen V, Hou X, Kaipiainen M, Vaaramaa K, Lindahl P, Rautio S, Kangas S, Jortikka S, Vasara L (2021) Intercomparison for separating Pu and Am from environmental samples by a joint radioanalytical method. NKS-442, Roskilde, Denmark. ISBN 978-87-7893-534-2. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214697264
Sidhu RS, Østmo K, Ruud AB (2004) Sequential extraction chromatographic separation of Pu, Am, Cm and Sr in soil, sediment and urine samples. In: Extraction chromatographic separation of Sr, Pu and Am in Environmental Samples’. Ph.D. Thesis Unipub AS, Oslo. ISSN 1501–7710
Sidhu RS (2004) Extraction chromatographic separation of Sr, Pu and Am in environmental samples. Ph.D. Thesis Unipub AS, Oslo. ISSN 1501–7710
Sidhu RS (2002) Sequential analysis of Sr, Pu, Am and Cm in low-level liquid radioactive effluents. Radioprot Colloq 37:917–920
Talvitie NA (1972) Electrodeposition of actinides for α-spectrometric determination. Anal Chem 44(2):280–283. https://doi.org/10.1021/ac60310a013
Wang Y, Hou X, Zhang W, Zhang L, Fan Y (2021) Determination of ultra-low 236U in environment samples using ICP-MS/MS measurement and chemical separation. Talanta 224:121882. https://doi.org/10.1016/j.talanta.2020.121882
International Standard ISO 9696:2017. Water quality – gross alpha activity – Test method using thick source. ISO, Geneva, Switzerland
International Standard ISO 11704:2018. Water quality – gross alpha and gross beta activity – Test method using liquid scintillation counting. ISO, Geneva, Switzerland
International Standard ISO 10704:2019. Water quality – gross alpha and gross beta activity – Test method using thin source deposit. ISO, Geneva, Switzerland
Bé M-M, Chisté V, Dulieu C, Mougeot X, Browne E, Chechev V, Kuzmenko N, Kondev F, Luca A, Galán M, Nichols AL, Arinc A, Huang X (2010) Table of Radionuclides (Volume 5 – A=22 to 244). BIPM Monographie BIPM-5, Bureau international des poids et mesures, Sèvres. https://www.bipm.org/en/publications/monographies
Paatero J (2000) Deposition of chernobyl-derived transuranium nuclides and short-lived radon-222 progeny in Finland. Finnish Meteorological Institute Contributions No. 28, Helsinki, pp 29. ISBN 951-697-522-4
Mandel J, Paule RC (1970) Interlaboratory evaluation of a material with unequal numbers of replicates. Anal Chem 42(11):1194–1197. https://doi.org/10.1021/ac60293a019
Paule RC, Mandel J (1982) Consensus values and weighting factors. J Res Natl Bur Stand 87(5):377–385. https://doi.org/10.6028/jres.087.022
Bé M-M, Chisté V, Dulieu C, Mougeot X, Chechev V, Kondev F, Nichols AL, Huang X, Wang B (2013) Table of Radionuclides (Volume 7 – A=14 to 224). BIPM Monographie BIPM-5, Bureau international des poids et mesures, Sèvres. https://www.bipm.org/en/publications/monographies
Hou X, Allard S, Hansson N, Hatakka A, Hovi P, Eriksson M, Olszewski G, Salminen-Paatero S, Bruzell F, Suutari T, Ekerljung L, Andersson A, Davidsson M, Öhlin H, Åberg H, Anderot M, Tovedal A, Rautio S, Kangas S (2019) Simultaneous determination of isotopes of Pu, Am and Cm in reactor water samples – Report of OptiMethod 2018 project. NKS-415, Roskilde, Denmark. ISBN 978–87–7893–504–5. https://www.nks.org/en/nks_reports/view_document.htm?id=111010214696185
Giménez I, Bagán H, Tarancón A (2023) Fast analysis of gross alpha with a new plastic scintillation resin. Anal Chim Acta 1248:340905. https://doi.org/10.1016/j.aca.2023.340905
Goldstein SJ, Hensley CA, Armenta CE, Peters RJ (1997) Environmental and human monitoring of Americium-241 utilizing extraction chromatography and α-spectrometry. Anal Chem 69(5):809–812. https://doi.org/10.1021/ac960728r
Hallstadius L (1984) A method for the electrodeposition of actinides. Nucl Instrum Methods Phys Res 223(2–3):266–267. https://doi.org/10.1016/0167-5087(84)90659-8
LaRosa JJ, Cooper EL, Ghods-Esphahani A, Jansta V, Makarewicz M, Shawky S, Vajda N (1992) Radiochemical methods used by the IAEA’s laboratories at Seibersdorf for the determination of 90Sr, 144Ce and Pu radionuclides in environmental samples collected for the international Chernobyl project. J Environ Radioact 17(2–3):183–209. https://doi.org/10.1016/0265-931X(92)90025-O
Sill CW, Williams RL (1981) Preparation of actinides for a spectrometry without electrodeposition. Anal Chem 53(3):412–415. https://doi.org/10.1021/ac00226a008
Nilsson H, Ramebäck H, Skålberg M (2001) An improved method for α-source preparation using neodymium fluoride coprecipitation. Nucl Inst Methods Phys Res A 462(3):397–404. https://doi.org/10.1016/S0168-9002(01)00184-X
Salminen-Paatero S, Hou X, Olszewski G, Ekerljung L, Tovedal A, Vesterlund A, Andersson A, Kangas S, Ramebäck H (2021) Analyzing alpha emitting isotopes of Pu, Am and Cm from NPP water samples: an intercomparison of Nordic radiochemical laboratories. J Radioanal Nucl Chem 329(3):1447–1458. https://doi.org/10.1007/s10967-021-07913-7
Kazi ZH, Cornett JR, Zhao X, Kieser L (2014) Americium and plutonium separation by extraction chromatography for determination by accelerator mass spectrometry. Anal Chim Acta 829:75–80. https://doi.org/10.1016/j.aca.2014.04.044
Luskus CA (1998) Electroplating versus microprecipitation of the actinides in alpha-spectroscopic analysis. J Radioanal Nucl Chem 234(1):287–292. https://doi.org/10.1007/BF02389787
Edwards KE (1968) Isotopic analysis of uranium in natural waters by alpha spectrometry. Geological Survey Water Supply Paper 1696-F. U.S. Department of Interior, United States Government Printing Office, Washington. https://doi.org/10.3133/wsp1696F
Sill CW (1977) Determination of thorium and uranium isotopes in ores and mill tailings by alpha spectrometry. Anal Chem 49(4):618–621. https://doi.org/10.1021/ac50012a028
Reis AS, Temba ESC, Kastner GF, Monteiro RPG (2011) Combined procedure using radiochemical separation of plutonium, americium and uranium radionuclides for alpha-spectrometry. J Radioanal Nucl Chem 287(2):567–572. https://doi.org/10.1007/s10967-010-0774-3
Taddei MHT, Vicente R, Marumo JT, Sakata SK, Terremoto LAA (2013) Determination of long-lived radionuclides in radioactive wastes from the IEA-R1 nuclear research reactor. J Radioanal Nucl Chem 295(2):951–957. https://doi.org/10.1007/s10967-012-1865-0
Tölgyesi S, Gresits I, Past T, Szabó L, Volent G, Pintér T (2002) Determination of alpha-emitting isotopes in radioactive wastes. J Radioanal Nucl Chem 254(2):357–361. https://doi.org/10.1023/A:1021692304088
Aggarwal SK (2016) Alpha-particle spectrometry for the determination of alpha emitting isotopes in nuclear, environmental and biological samples: past, present and future. Anal Methods 8(27):5353–5371. https://doi.org/10.1039/C6AY00920D
Diez-Fernández S, Isnard H, Nonell A, Bresson C, Chartier F (2020) Radionuclide analysis using collision–reaction cell ICP-MS technology: a review. J Anal At Spectrom 35:2793–2819. https://doi.org/10.1039/D0JA00211A
Jaegler H, Gourgiotis A, Steier P, Golser R, Diez O, Cazala C (2020) Pushing limits of ICP-MS/MS for the determination of ultra-low 236U/238U isotope ratios. Anal Chem 92(11):7869–7876. https://doi.org/10.1021/acs.analchem.0c01121
Rodriguez M, Gascón JL, Suárez JA (1997) Study of the interferences in the determination of Pu, Am and Cm in radioactive waste by extraction chromatography. Talanta 45:181–187. https://doi.org/10.1016/S0039-9140(97)00118-5
Tavčar P, Smodišs B, Benedik L (2007) Radiological characterization of low-and intermediate-level radioactive wastes. J Radioanal Nucl Chem 273(3):593–596. https://doi.org/10.1007/s10967-007-0916-4
Gascón JL, Aceña ML, Suárez JA, Rodríguez M (1994) Radiochemical methods for the determination of plutonium, americium and curium in typical waste streams. J Alloy Compd 213–214:557–559. https://doi.org/10.1016/0925-8388(94)90987-3
Lee MH, Jeon YS, Song K (2009) Determination of activity concentrations and activity ratios of plutonium, americium and curium isotopes in radioactive waste samples. J Radioanal Nucl Chem 280:457–465. https://doi.org/10.1007/s10967-008-7442-x
Gorietti D, Giardina I, Arginelli D, and.Battisti, P. (2017) Determination of plutonium, americium and curium isotopes in radioactive metal wastes deriving from nuclear decommissioning. J Radioanal Nucl Chem 314(3):1785–1792. https://doi.org/10.1007/s10967-017-5553-y
Groska J, Vajda N, Molnár ZE, Bokori E, Szeredy P, Zagyvai M (2016) Determination of actinides in radioactive waste after separation on a single DGA resin column. J Radioanal Nucl Chem 309(3):1145–1158. https://doi.org/10.1007/s10967-016-4729-1
Dewberry RA, Boyce WT (1999) Plutonium measurements in Hanford DOE site waste samples. J Radioanal Nucl Chem 240(2):643–650. https://doi.org/10.1007/BF02349426
Jäggi M, Röllin S, Corcho Alvarado JA, Eikenberg J (2012) Determination of 241Pu in nuclear waste slurries: a comparative study using LSC and ICP-MS. Appl Radiat Isot 70(2):360–364. https://doi.org/10.1016/j.apradiso.2011.10.005
Choppin GR, Bond AH, Hromadka PM (1997) Redox speciation of plutonium. J Radioanal Nucl Chem 219(2):203–210. https://doi.org/10.1007/BF02038501
Salminen-Paatero S, Paatero J (2020) Separation method for Pu, Am and Sr in large air filter sample sets. MethodsX 7:100910. https://doi.org/10.1016/j.mex.2020.100910
Eichrom Technologies (2014) Americium, neptunium, plutonium, thorium, curium, Uranium, and strontium in Water, Analytical Procedure, Method No ACW17VBS, revision 1.2. https://www.eichrom.com/wp-content/uploads/2018/02/acw17-12_am-pu-u-np-th-sr-water-vbs.pdf
Bé M-M, Chisté V, Dulieu C, Browne E, Chechev V, Kuzmenko N, Kondev F, Luca A, Galán M, Pearce A, Huang X (2008) Table of Radionuclides (Volume 4 – A=22 to 244). BIPM Monographie BIPM-5, Bureau international des poids et mesures, Sèvres. https://www.bipm.org/en/publications/monographies
Gourgiotis A, Granet M, Isnard H, Nonell A, Gautier C, Stadelmann G, Aubert M, Durand D, Legand S, Chartier F (2010) Simultaneous uranium/plutonium separation and direct isotope ratio measurements by using CO2 as the gas in a collision/reaction cell based MC-ICPMS. J Anal At Spectrom 25:1939–1945. https://doi.org/10.1039/C0JA00092B
Rozas S, Herranz M, Idoeta R (2021) Uncertainty and detection limits of 241Pu determination by liquid scintillation counting (LSC). Appl Radiat Isot 176:109906. https://doi.org/10.1016/j.apradiso.2021.109906
Habibi A, Le Corre R, Wampach Aubert C, Fleury S, Hennequet P, Augeray C, Galliez K, Agarande M (2021) Quantification of curium isotopes in environmental samples: drawbacks, speciation and specific tracer. J Radioanal Nucl Chem 329(2):545–554. https://doi.org/10.1007/s10967-021-07751-7
Mitchell PI, Holm E, León Vintró L, Condren OM, Roos P (1998) Determination of the 243Cm/244Cm ratio alpha spectrometry and spectral deconvolution in environmental samples exposed to discharges from the nuclear fuel cycle. Appl Radiat Isot 49(9–11):1283–1288. https://doi.org/10.1016/S0969-8043(97)10061-6
Horwitz EP, Chiarizia R, Dietz ML, Diamond H, Nelson DM (1993) Separation and preconcentration of actinides from acidic media by extraction chromatography. Anal Chim Acta 281(2):361–372. https://doi.org/10.1016/0003-2670(93)85194-O
Gharibyan N, Dailey A, McLain DR, Bond EM, Moody WA, Happel S, Sudowe R (2014) Extraction behavior of americium and curium on selected extraction chromatography resins from pure acidic matrices. Solvent Extr Ion Exch 32(4):391–407. https://doi.org/10.1080/07366299.2014.884888
Ham GJ (1995) Determination of actinides in environmental materials using extraction chromatography. Sci Total Environ 173(174):19–22. https://doi.org/10.1016/0048-9697(95)04746-8
Martin JP, Odell KJ (1997). The development of emergency radioanalytical techniques for the determination of radiostrontium and transuranic radioisotopes in environmental materials. In: Rapid radioactivity measurements in emergency and routine situations, National Physical Laboratory, Teddington, UK, pp 101–113
Crable A, Gharibyan N, Capote A, Bombard A, Happel S, Maddigan G, Sudowe R (2009) Absorption study of Am and Cm to TEVA, TRU and DGA resins. In: NKS-B workshop on radioanalytical chemistry, NKS-218, Roskilde (Denmark). https://www.nks.org/en/nks_reports/view_document.htm?id=111010111120204
Hou X (2018) Liquid scintillation counting for determination of radionuclides in environmental and nuclear application. J Radioanal Nucl Chem 318(3):1597–1628. https://doi.org/10.1007/s10967-018-6258-6
Jobbágy V, Wätjen U, Merešova J (2010) Current status of gross alpha/beta activity analysis in water samples: a short overview of methods. J Radioanal Nucl Chem 286(2):393–399. https://doi.org/10.1007/s10967-010-0709-z
Palomo M, Villa M, Casacuberta N, Peñalver A, Borrull F, Aguilar C (2011) Evaluation of different parameters affecting the liquid scintillation spectrometry measurement of gross alpha and beta index in water samples. Appl Radiat Isot 69(9):1274–1281. https://doi.org/10.1016/j.apradiso.2011.04.020
Monroy-Guzman F, Lopez Sibaja Y, Javier Galicia Caballero F (2020) Thin source spraying method for gross alpha/beta measurements with emphasis on alpha/beta particle self-absorption. Appl Radiat Isot 164:109320. https://doi.org/10.1016/j.apradiso.2020.109320
Jobbàgy J, Merešová UW (2014) Critical remarks on gross alpha/beta activity analysis in drinking waters: conclusions from a European interlaboratory comparison. Appl Radiat Isot 87:429–434. https://doi.org/10.1016/j.apradiso.2013.11.073
Stojković; I., Tenjović, B., Nikolov, J, Todorovi, N. (2017) Possibilities and limitations of color quench correction methods for gross alpha/beta measurements. Appl Radiat Isot 122:164–173. https://doi.org/10.1016/j.apradiso.2017.01.041
Dobrin RI, Dulama CN, Ciocirlan CN, Toma AL, Stoica SM, Valeca M (2013). Alpha activity measruement with LSC, Institute for Nuclear Research, Pitestii, Romania, NUCLEAR-2013. ISSN 2066–2955
Mirashi NN, Chander K, Aggarwal SK (2000) Liquid scintillation counting techniques for the determination of some alpha emitting actinides: aA review. Fuel Chem Div, Bhabha Atomic Research Center, Mumbai, 2000, BARC/2000/E/037
Piraner O, Jones RL (2021) Universal use of alpha/beta mode in liquid scintillation counting analysis for both alpha/beta and single nuclide determination. J Radioanal Nucl Chem 327(2):975–983. https://doi.org/10.1007/s10967-020-07557-z
Fons-Castells J, Tent-Petrus J, Llauradó M (2017) Simultanous determination of specific alpha and beta emitters by LSC-PLS in water samples. J Environ Radioact 166:195–201. https://doi.org/10.1016/j.jenvrad.2016.04.035
Hou X, Zhang W, Wang Y (2019) Determination of femtogram-level plutonium isotopes in environmental and forensic samples with high-level uranium using chemical separation and ICP-MS/MS measurement. Anal Chem 91(18):11553–11561. https://doi.org/10.1021/acs.analchem.9b01347
Acknowledgements
The authors would like to thank the Nordic Nuclear Research NKS-B programme (www.nks.org) for funding the RESINA project. The authors would also like to thank the other participating laboratories, namely Technical University of Denmark, Institute for Energy Technology Kjeller, Taiwan Power Company Radiation Laboratory and UK National Nuclear Laboratory for provision of data and collaboration. National fundings were given by Finnish Research Programme on Nuclear Waste Management KYT 2022. A-LABOS-EX-PR-SC-01 project is thanked for the CEA self-funding. CEA LASE laboratory operators Yousim Taing, and Jacques Bubendorff are thanked for their experimental contributions. Hidex customer support personnel is thanked for the help in interpretation of the LSC spectra. Alex Sanz, Isaac Guerra and Hector Navarro (Universitat de Barcelona) are thanked for the collaboration and provision of the α-PSresin.
Funding
Open Access funding provided by Technical Research Centre of Finland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leskinen, A., Jerome, S., Lavonen, T. et al. Intercomparison exercise on difficult to measure alpha radionuclides in spent ion exchange resin. J Radioanal Nucl Chem 333, 563–584 (2024). https://doi.org/10.1007/s10967-023-09233-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-023-09233-4