
Abstract
Characterisation of activated reactor components is a multifaceted effort, which include both experimental work and activation calculations which together form a comprehensive characterisation approach. However, both the experimental work and activation calculations have their constraints and uncertainties. This work presents the characterisation of a highly activated stainless steel pipe including sampling planning, sampling, sub-sampling, elemental and radionuclide analyses, and activation calculations. 55Fe, 63Ni and 60Co analyses were carried out in a trilateral intercomparison exercise and the results were compared with activation calculations. The results showed excellent alignment between experimental and activation calculation results.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Characterisation of high activity materials is a challenge as both the representativeness of the sample and radiation safety must be considered. Small sample sizes are preferred when handling high activity materials whereas large sample sizes are preferred especially with heterogenous materials. In the case of activated reactor components, homogenisation is not possible (i.e., solid materials with activation gradients) and consequently a compromise is needed to collect a sample as representative as possible.
The two main materials contributing to radioactive decommissioning waste volumes are the biological shield concrete and the reactor core structure steels. Steel contains at least 50% iron, but various amounts of alloying elements have been added to the different steel types to enhance one or several specific properties, e.g., strength, hardness, corrosion resistance or machinability [1]. The alloying elements generally include carbon, manganese, silicon, nickel, chromium, molybdenum, vanadium, titanium, niobium, and aluminium [1]. The roles of the alloying elements are shown in Table 1. Overall, there are thousands of different steels and consequently their classification is difficult [1]. However, steels are generally classified as carbon steel and alloy steel [1]. The carbon steels, which are also called plain-carbon steels, constitute a family of iron-carbon-manganese alloys [1]. The alloy steels are alloys of iron with the addition of one or more of the following elements: C, Mn, Si, Ni, Cr, Mo, and V [1]. The alloy steels cover a wide range of steels, including low-alloy steels, stainless steels, heat-resistant steels, and tool steels [1]. Stainless steels are corrosion-resistant steels that contain at least 10.5% chromium, which forms a passive layer on the steel surface protecting it from corrosion [1]. The stainless steels are divided to five types, i.e., austenitic, ferritic, duplex, martensitic, and precipitation-hardening steels [1]. In general, the austenitic steels contain 16–26% Cr and 6–22% Ni [1]. The ferritic stainless steels are iron-chromium alloys with Cr content between 10.5% and 27% and without Ni or in minor amounts, i.e., less than 1% [1]. The dublex stainless steels are approximately equal mixtures of ferrite and austenite. Martensitic stainless steels contain added C allowing higher Cr content [1]. Precipitation-hardening stainless steels are iron-chromium-nickel alloys which have high strength and toughness through addition of Al, Ti, Nb, V and/or N [1]. Other stainless steels, such as tool steels, are alloy steels that are used for cutting or machining of other materials and they contain various levels of Cr, Ni, Mo, W, V, and Co [1]. In nuclear applications, carbon steel is typically used in massive structures as a radiation shield outside the reactor tank. For example, type 304 austenitic stainless steel, which is typically used in reactor core structures due to its mechanical and chemical properties, contains 18–20% Cr and 8–12% Ni. In some components, acid resistant stainless steel (containing as well 2–3% Mo) may also be used.
For all activated steel types, the main gamma emitters are 60Co (t1/2 5.27 a), 54Mn (t1/2 312 d), and 51Cr (t1/2 27.7 d). Stainless steel, which is typically used in reactor core structures due to its mechanical and chemical properties, contains after irradiation the same radionuclides as carbon steel, e.g., 60Co, 14C and 55Fe. However, due to the much higher nickel and chromium content in stainless steel, the 63Ni, 59Ni and 51Cr activities are much higher compared to ones present in carbon steel.
Activation calculation is an advantageous tool to carry out assessments of activation levels for reactor components such as steel components. Preliminary calculations are often carried out conservatively due to a lack of information on detailed chemical compositions, irradiation history, and neutron fluxes. Nonetheless, the activation calculations can give indicative estimations on the activation level of the radionuclides and consequently dose rates can be estimated. This information is of great value during the sampling plan so the analyses can be prepared while keeping in mind the goal i.e., a safe and representative sampling to minimise the need for a secondary sampling campaign and consequently lower the use of resources. After validation of the method by comparing the results of the activation calculation with the measurements performed in the laboratory, the calculations can be used on larger waste volumes and different reactor components with varying activation levels. Consequently, the cost of characterisation is reduced.
The radionuclides in radioactive waste are divided into easy to measure (ETM) and difficult to measure (DTM) radionuclides. The ETM radionuclides (i.e., gamma emitters) can be measured non-destructively with short radiation exposure time to the operator whereas the DTM radionuclides (i.e., alpha and beta emitters) require hands-on analysis lasting up to several days. Therefore, a careful assessment of the activity levels (i.e., the amount of sample needed for detecting radionuclides of interest), representativeness, and radiation exposure times needs to be carried out case by case. The ETM analysis is a well-established technique measuring discrete gamma emissions resulting in quantitative analyses enabled by geometry and efficiency calibrations. Most often the gamma emitters can be determined directly from a sample, without a need for a preceding radiochemical separation procedure. However, sample preparations can include homogenisation and placement in well-defined geometries.
Radiochemical analysis requires separation and purification of the DTM of interest from the interfering radionuclides and the stable elements using destructive treatments (e.g., acid digestion, alkali fusion), precipitations, ion exchange, and chromatographic resins prior to detection with liquid scintillation counting or alpha spectrometry. Radiochemical procedures of DTM analyses in activated steels have been reported in the literature [2,3,4]. However, reporting of DTM analyses in highly activated steels is scarce. For example, Corcho-Alvarado et al. [5] reported DTM analysis of a 1 mg steel sample, which had been taken from the bottom structure of a nuclear fuel element with 60Co activity concentration of 3.0 × 108 Bq g−1 [5]. The 1 mg sample (dose rate 0.3 mSv h−1) was dissolved and diluted into four aliquots that were taken for DTM analyses [5]. The averaged DTM results in each aliquot was reported to have excellent repeatability, i.e., 1030 ± 20 Bq ml−1 of 55Fe and 320 ± 33 Bq ml−1 of 63Ni [5]. Even though Corcho-Alvarado et al. [5] did not discuss the sampling procedure nor the reason for the small sample mass, it can be speculated that the radiation safety of the analyser played a role. Additionally, the representativeness of the sample was not discussed. The radiochemical procedures do not necessarily differ between low, medium and high-activity materials, and also radiation dose can be lowered with exposure time, distance from the source, and shielding. However, the practical limitations in the radiochemical analyses (i.e., hands-on working in a fume cupboard, risks associated with destructive use of acids; and possible spills due to obstructive shielding) may dictate that a compromise is needed between radiation protection and chemical safety. Optimisation of sampling and sub-sampling as well as careful planning beforehand are therefore essential in the sampling of highly radioactive materials.
Activation is dependent on the chemical composition of the material, the irradiation history, and the neutron flux. The activation calculations are directly dependent on the above-mentioned parameters. The minor impurities present in the material are often the major constituent in activation, e.g., Co and Ni, but also Mo, Se, Ho, etc. In some cases, the material specifications do not include all the minor impurities, or only a limit of detection is reported. Therefore, there is a need to carry out chemical composition determinations, which can be carried out using destructive and non-destructive methods. A common destructive method (destructive analysis, DA) is to mineralise the material using mixtures of acids and measure the chemical composition of targeted elements (i.e., relevant radionuclide precursors such as Co and Ni) using optical or mass spectrometric technologies such as inductively coupled plasma optical emission spectrometer (ICP-OES) or inductively coupled plasma mass spectrometer (ICP-MS). A semi-destructive method (semi-destructive analysis, semi-DA) to determine elemental mass-percentages is glow discharge optical emission spectrometer (GD-OES). In this case, the material surface is studied for the chemical composition. A thin layer of the material surface is destroyed in each measurement. One benefit of GD-OES over ICP-MS and ICP-OES is in the analysis of C which is one precursor for long-living 14C. ICP-MS and ICP-OES are not suitable for measurement of C due to weak ionisation of C in ICP, being only 1–5% [6, 7]. In addition, ICP measurements most often require Ar gas, which contains C as an impurity, as well as many common reagents contain C. Neutron activation analysis (NAA) is an example of non-destructive chemical analysis (non-destructive analysis, NDA). However, the availability of NAA is much more limited compared to widely available ICP-OES and ICP-MS technologies.
As in any analysis, method validation is needed for both ETM and DTM analyses. Especially for DTM analyses, there are no commercially available reference materials against which laboratories could assess their radioanalytical performances. In order to fill this gap, intercomparison exercises have been carried out for both spiked and real materials [2, 4, 8,9,10,11,12,13]. The benefit in analysis of real materials is associated with real difficulties in the analyses. For example, a difference in radioanalytical performance was found between spiked and real sample analyses in a previous intercomparison exercise, where 63Ni was analysed from spiked water and reactor process water samples [8]. Real sample analyses may produce more scattered results than spiked sample analyses, due to impurities (e.g., 60Co in steel) or interfering bulk elements (e.g., Ca in concrete) in the real material and impurity-induced challenges for the radioanalytical separation method. Moreover, the speciation of DTMs may be significantly different in contaminated and activated waste, and mutually different radioanalytical separation procedures for these waste types might be then necessary [2].
This paper describes the sampling plan and execution, characterisation (i.e., activation calculations and elemental, ETM and DTM analyses), method validation and lessons learned in analysis of a high activity stainless-steel sample. The studied sample originated from FiR1 TRIGA Mark II type research reactor, which is currently under decommissioning. The sampling plan and execution considered the characterisation needs (i.e., representativeness of the sampling and formation of a scaling factor) and needs in the analyses (i.e., radiation and chemical safety and sample masses). Elemental analyses on inactive and low activity samples were carried out in order to feed input in the activation calculations. A trilateral intercomparison exercise was organised to validate ETM (i.e., 60Co) and DTM (i.e., 55Fe, and 63Ni) analyses. All analysis results were compared with the activation calculations.
Characterisation in FiR1 decommissioning project
FiR1 research reactor was the first nuclear reactor in Finland with initial power of 100 kW when inaugurated in 1962. The power was raised to 250 kW in 1967 and a boron neutron capture therapy (BNCT) was started in mid 1990s, which included both changes in the reactor structures and operation. During 1962–2015 FiR1 research reactor has been used for education, research, isotope production and other services utilising neutrons. Characterisation of the reactor’s activated structures has been an ongoing effort since the permanent shutdown in 2015 [14,15,16]. Both activation calculations and experimental studies have been utilised to form material-specific scaling factors prior to dismantling. Radiochemical methods have been developed and validated in several intercomparison exercises [2,3,4, 9,10,11,12,13] whereas this paper presents characterisation of the first sample representing highly activated FiR1 materials, namely a cable pipe for stainless steel cladded instrumented fuel element. Previously established sampling and pre-treatment methods were utilised throughout the work, keeping the radiation safety of the workers in the forefront without forgetting analytical objectives.
FiR1 stainless steel
The TRIGA research reactor core is approximately 46 cm in diameter and has 89 fuel element locations in five circular rings. Two of the fuel elements were instrumented, i.e., they had a thermocouple built inside of them to monitor the fuel temperature (Fig. 1). The signal from the thermocouple instrument was led to the surface of the reactor tank inside the stainless-steel tubing. The material specifications indicated that the instrumentation pipes were made of stainless steel by General Atomics. However, no detailed specifications on chemical compositions were available. The stainless steel pipes were connected to instrumented fuel elements, which were similar to other fuel elements but with incorporated temperature probes. Because the lower end of the cable pipe was directly above the reactor core, it was highly activated.
The spent nuclear fuel from the FiR1 reactor was removed in 2020 and the instrumentation pipes were cut prior to packaging the fuel. Approximately 5 cm of the instrumental pipes remained with the fuel elements. A maximum of 100 cm of the activated ends of the pipes (corroded instrument cables were left inside) were placed in a shielded dry storage. The remaining lengths of the pipes (i.e., over 500 cm) were not activated and were handled as free-release waste.

TRIGA research reactor instrumented fuel elements. The samples studied in this article were collected from the lead-out tubing
Experimental
The experimental work was a multifaceted study including elemental, ETM and DTM analyses, corresponding sample preparations (i.e., sub-sampling, pre-treatment and analysis), activation calculations, trilateral intercomparison exercise on characterisation of DTM and ETM radionuclides, and comparison of the measurement results with activation calculations. The overarching theme throughout the study was sample representativeness, radiation safety and efficient use of resources. The sub-sampling, elemental analyses and initial gamma spectrometric analyses were carried out at Centre of Nuclear Safety (CNS) at VTT. The elemental analysis results fed information into the planning of DTM analyses (i.e., solubility and Fe and Ni compositions) and activation calculations.
Sampling
The inactive, low and high activity locations of the FiR1 stainless-steel instrumentation pipe were manually sampled using a manual 3–45 mm pipe cutter, with which the cutter blade was pressed on the pipe and rotated around the pipe until break through. Inactive samples were collected from the non-activated upper part of the 5 m long pipe. The samples were labelled as sample A (inactive), B (low activity) and C (high activity).
The inner and outer diameters of the pipe were easily determined (i.e., without exposure to radiation) from the inactive stainless-steel sample A using a vernier calliper. The lengths and masses of samples A and B were measured using the vernier calliper and an analytical balance. A close inspection of samples A and B showed that there was some rust in the inner wall of the pipe. The rust originated from rusted instrumental wires, which run throughout the pipe. Therefore, it was very possible that the highly active sample C would also contain rust in the inner wall and consequently, it should be removed prior to ETM and DTM characterisation. The height of sample C was measured inside a hot cell using OGP Flash CNC 200 optical measurement device.
Sub-sampling
Sub-samplings of the stainless-steel samples A, B and C were needed for the elemental, ETM and DTM analyses. The material was hard and the pipe wall was relatively thick (2.4 mm) with a smooth surface and therefore sub-samplings by hand (e.g., file, pliers, saw) was considered to be an inefficient technique. Hand operated pliers would have not had enough strength to cut small pieces. Sawing would have suffered from precision due to the smooth surface and additionally causing contamination of the equipment and possible air contamination. Similarly, filing would have also caused contamination of the equipment and possible air contamination. A wire EDM (Electrical Discharge Machining) is a powerful and very precise tool for cutting different objects. VTT has a GF AgieCharmilles Cut 200 mS wire EDM inside a hot cell and it has been utilised for cutting specimens for mechanical testing. For example, cutting Charpy impact, tensile and miniature compact tension specimens was performed with the wire EDM related to the investigation of decommissioned Swedish Barsebäck 2 reactor pressure vessel weld materials [17]. Wire EDM has also been used for preparing samples for hardness testing and Charpy specimens cross-section evaluation for finding the location of the fracture initiation [18]. Wire EDM can cut only electrically conductive materials. The maximum dimensions for the wire EDM workpiece are 1000 mm (length), 550 mm (width) and 220 mm (height) and axis travel lengths are 350 mm (X-axis) 220 mm (Y-axis) and 220 mm (Z-axis). However, the hot cell environment has restrictions for workpiece size e.g., the lift capacity of a small crane in the hot cell is 100 kg and for the A200 series manipulators by Wälischmiller Engineering, that are used for the remote handling of items inside the hot cell, it is theoretically 8 kg.
The wire EDM operates underwater and produces contaminated water which is purified with mechanical cartridge filters. Before use, the water is deionized with resin to reduce the electrical conductivity of the water. Deionising resin and used wire are contaminated when radioactive objects are cut. In general, 760 l of water circulates through the wire EDM and approximately 10 m of contaminated wire is produced per minute of cutting. Additionally, in each cut the wire releases approximately 0.4 mm width of material along the cut, depending on material height and surface quality requirements. Wire EDM produces much less material waste compared to conventional machining methods and that is a clear advantage in perspective of radioactive waste handling and unique material cutting. During the project, only inactive samples were allowed to be handled with the wire EDM and therefore, the inactive sample A was sub-sampled to demonstrate the wire EDM’s capabilities and study the behaviour of the material (e.g., the thinness of the slices, cutting configuration) for future applications.
Figure 2 illustrates the EDM cut model, which was done with IronCAD software. The cutting path of the wire EDM was programmed with PEPS CAM software, which enabled the cutting of a thin slice into four pieces, which would remain attached to the pipe and could be safely detached using a manipulator. The depth of the cut was planned to result in sub-samples of 100–200 mg, which was considered to be a good size for a high-activity sample sub-sampling i.e., enough material for the ETM and DTM measurements with relatively low dose rate.

Inactive sample A with 32 mm height (a), CAD-model for the EDM cut for 32 mm height inactive sample A (b), EMD cut sample A pieces (c), decontaminated high activity sample C pieces (approximately 0.1 g) cut with milling machine (d) schematic presentation of hydraulic cutter sub-sampling (e), overall schematic presentation of the sample A, B and C locations (not in scale) (f)
Sub-sampling of the highly activated sample C was needed to minimize exposure to external radiation during the radiochemical analyses. Since the wire EDM was not an option at this point, sub-sampling was carried out with a MeteCNC 3-axis milling machine which is used for opening irradiation capsules, for example. CNC milling machine operates with 3 axes which travel distances of 675 mm, 390 mm, and 350 mm (X, Y, Z-axes) and it includes an automatic magazine for 7 tools. The machine was tailor-made for hot cell use and all the functionalities can be operated remotely or from the maintenance room on the back of the machine to minimise the need to go inside the hot cell. Cutting with a milling machine produces a lot of small chips and to protect those spreading throughout the cell the cutting area was protected with movable plastic covers. After cutting and collecting the samples most of the chips were cleaned with a squeegee attached to a manipulator and the rest of the chips were removed with in-cell vacuum cleaner. The methodology was first tested inside a hot cell with the low activity sample B (Fig. 2) to optimise the cutting parameters. The determining factor of the parameters was the form of the sub-samples. Optimised parameters were spindle speed 800 rev min−1, feed rate 0.35 rev min−1 and the depth of cut 2.0 mm. Milling was performed with a 20 mm indexable end mill cutting tool.
The lessons learned were directly utilised in the sub-sampling of the highly activated sample C. The sample was placed inside a plastic vial to facilitate easy handling of the sample. Actually, handling of the high-activity sample C was planned to start from packing the sample inside a transport casket at the FiR1 research reactor site for transportation to CNS. The sample C inside the plastic vial was placed in a metal jar with an upright handle and then placed inside a transport casket. The upright handle enabled easy lifting of the jar from the transport casket with manipulators inside the hot cells. Additionally, the type of plastic vial had already been successfully handled with manipulators (e.g., opening and closing of the cap). These considerations become relevant when handling of the samples needs to be carried out with manipulators.
Elemental analyses
Elemental analyses were carried out for inactive and low-activity sub-samples, which were decontaminated with HNO3 prior to analyses. Both destructive ICP-OES (Agilent 5100 SVDV) and ICP-MS (ThermoScientific Element 2) techniques require the dissolution of the solid sample prior to analysis. The sample size does not have significant restrictions as long as the sample is soluble. A literature study showed that the solubility of stainless steel should not be problematic as different acid mixtures (e.g., aqua regia, aqua regia combined with HF) had been reported successful [19,20,21,22]. However, different types of stainless steels, and steels in general, can behave differently and therefore, the inactive sub-samples were also used to test the solubility of the studied stainless steel. The measured elements (e.g., Fe, Ni, Co) were selected in order to provide input data for the activation calculations, categorisation of the steel type, and complementary data for the design of the DTM analyses.
The GD-OES (Spectruma Analytik GMBH α GDA) measurements were focused on the analysis of C, which is one precursor for 14C activation. Additionally, there was an interest to compare the ICP-MS and ICP-OES results with the GD-OES results, because the latter technique gives the results in mass percentages and the measured elements are selected based on the main elements that should be found in the material. The GD-OES analysis is a spectroscopic method, where a high direct voltage is applied between an anode with a 2.5 mm slit and the sample that acts as a cathode. In low pressure argon gas, the method is based on sputtering technique, as the argon cations knock out the sample surface atoms. The sample atoms are transferred to the detector for the characteristic wavelength of the sample atoms to be analysed. The chemical composition can be determined by comparing the wavelengths to a set of reference materials with similar compositions.
N analyses (Bruker G8 Galileo) were carried out as N is the main precursor for 14C activation. The N content in the steel sample was analysed using hot melt extraction method on a gas analyser using a thermal conductivity detector. The sample material was cut into 0.1 g chips and three repetitions were measured. The samples were melted in an impulse furnace in a graphite crucible between two electrodes, heating up to 2680 K, extracting the nitrogen gas detected using argon as inert carrier gas.
Gamma spectrometric measurements at VTT
Gamma spectrometric measurements were carried out using two high-purity Ge-detectors. Detector 1 (coaxial, 20% relative efficiency, high purity Germanium by Canberra) was located in a hot cell, in which gamma activities of arriving and departing transports are carried out. Detector 2 (p-type, 18% relative efficiency, high purity Germanium by Canberra) was reserved for characterisation of laboratory samples. The low activity sample B (23 mm height piece of pipe, see Fig. 2) was measured with both detectors using at least overnight measurement time. The high activity sample C (33.9 mm height piece of pipe, see Fig. 2) was measured only with Detector 1 inside the hot cell whereas sub-samples of sample C were measured with Detector 2 (measurement time 500–700 s). The measurements of samples B and C using both detectors were carried out in order to compare the obtained activity results. Sample B was measured in the hot cell using Detector 1 at 700 mm distance (0.5% dead time) and sample C at 795 mm distance (6% dead-time). Sample B was measured at 40 mm distance (0.05% dead time) and sub-samples of sample C (approximately 0.1 g of cut pieces, see Fig. 2) were measured at 120 mm distance (approximately 2% dead time) using Detector 2. Measurements of the sample C sub-samples were also carried out to determine their homogeneity, which was a needed parameter in the intercomparison exercise.
Trilateral intercomparison exercise
Method validation of the DTM analyses is challenging since there are no commercially available DTM reference materials. Spiking of an inactive material with DTM standard solutions is one way to validate the radiochemical methods. However, spiked materials may not include the difficulties encountered when analysing real materials, and therefore, an intercomparison exercise, in which real materials are studied, is a highly beneficial way to validate the radiochemical analysis methods. Even though ETM analyses are generally reported to be easy to perform, intercomparison exercises are also highly beneficial for gaining confidence. Therefore, an intercomparison exercise on ETM and DTM analyses was carried out by three laboratories. Each laboratory received approximately 100 mg of high-activity stainless-steel sub-samples and the homogeneity was determined using gamma spectrometry as previously explained. The selected ETM was 60Co and the selected DTMs were 55Fe and 63Ni. The analysis results were statistically analysed according to ISO 13,528 standard [23]. The same methodology was followed as in previous exercises [2, 4, 9,10,11,12,13] i.e., robust means and robust standard deviations were calculated using Algorithm A. which is robust for outliers up to 20% [23]. Performance assessments were carried out using z score of Eq. (1). which is a recommended method in cases where the assigned value xpt is calculated from the submitted results [23]. The submitted results (noted xi) were assessed against the assigned values derived from the participants’ results [23]. The z score results were acceptable when |z| ≤ 2.0. a warning signal was given for results with 2.0 < |z| < 3.0. and |z ≥ 3.0 results were unacceptable [23]. The assigned values were also compared with activation calculation results.
where xi = the value given by a participant i. xpt = the assigned value. σpt = standard deviation for the proficiency assessment.
Activation calculation
Estimating the activation reactions in the reactor structures was a two-stage process. First, a particle transport code was used to solve the neutron fluxes inside the reactor structures and components, and then this data was used in a point-depletion code, which took the energy dependent neutron flux values from the transport calculations together with the material composition data and operating history to determine the quantity of neutron activation products. This study applied Monte Carlo based neutron transport code MCNP and a point-depletion code ORIGEN-S [24, 25]. The procedure utilised is presented in Fig. 3 [26].

Overview of the applied calculation steps [26]
The instrumentation pipe studied in this article had been used in the FiR1 reactor tank for years 1987–2015. There have been changes in the other reactor structures during those years, but their effect on the core and instrumented fuel elements was considered negligible. Therefore, only one neutron spectrum calculated with MCNP code was used. However, neutron flux decreases rapidly directly above the core and consequently, the results were calculated to cover 1 cm increments in the 0–12 cm above the core. Calculated thermal neutron flux densities are presented in Table 2.
The operating history in a research reactor is very complicated with typically hundreds of start-ups and annual shutdowns. For simplification, it was assumed that all the operating hours of the reactor each year have been accumulated in a single run and the reactor has been shut down for the rest of the year. Annual operating hours were manually collected from reactor operating diaries. In total, the instrumentation tube has been exposed to neutron irradiation for 18 998 h.
Measured stainless steel composition was assumed in the calculation and ENDF/B-VI formatted cross sections were used for the activation reactions.
Results and discussion
Sampling and sub-sampling results
Sampling of the stainless-steel samples using the manual cutter was quick and easy. The minimum height of a sample was practically approximately 2 to 3 cm and therefore, sub-samplings were required. The several sub-samplings of the inactive and low-activity samples were carried out to prepare appropriately sized measurement targets for the elemental analyses and to practice sub-sampling of the high activity sample (i.e., CNC milling machine). The sub-sampling using the EDM was demonstrated to be a very precise tool and the cutting parameters were successfully designed for a highly active sample even if the technique was utilised only for the inactive sample at this point. However, the desired parameters especially for the GD-OES (i.e., flat and smooth surface) and gas analyser (i.e., 5 × 5 × 5 mm pieces of approximately 0.5–1 g) could have been catered using the EDM if the parameters would have been known at the beginning of the project. This realisation resulted in the first lesson learned: the sampling and sub-sampling plan to cater several different types of characterisation techniques (i.e., NDA, DA and semi-DA) should be established to great detail in order to prepare appropriate sub-samples with minimum effort and resources.
The radiation dose to the operator’s hands during the sampling of the high activity sample C was estimated to be low, i.e., < 4 µSv. However, a significant amount of effort was required for the sub-sampling. The use of EDM would have been a relatively quick technique (i.e., half a day) but such a high precision tool is not necessary in destructive analyses. Additionally, EDM would have produced secondary waste, namely contaminated resin and wire. The use of CNC milling machine was able to produce small shavings of sub-samples as demonstrated with the low activity sample. Collection of the shavings using the manipulators was relatively easy for the experienced operator. However, a major drawback was realised during the sub-sampling of low activity sample B, as contamination (characterised by presence of 54Mn) originating from a previous campaign was measured in the sub-samples when they were taken out of the hot cells. Consequently, decontamination of the sub-samples was especially important. As the major gamma emitters of the previous campaign were 54Mn and 60Co and the studied samples contained only 60Co, the presence of only 60Co in the decontaminated sub-samples was an indicator of successful decontamination.
The radiation safety during the sampling and the significant work carried out for the sub-sampling of the high activity sample C, were a driving force to establish a better procedure in a proceeding sampling campaign. Even if the estimated dose to the operator’s hands was relatively low, it did not follow the As-Low-As-Reasonably achievable (ALARA) principle. An increase of even a few cm distances between the pipe and hands would lower the dose rate significantly. Therefore, the sampling procedure in the proceeding sampling campaign was updated to include a hydraulic cutter (Cembre B-TC550 by Teleinstrument) and tongs, which enabled several cm of distance from the pipe to the hands. Additionally, the pipe was shielded with lead bricks and only few centimetres of the most active end of the pipe was exposed during sampling. The benefit of using a manual cutter compared to the hydraulic cutter is that the pipe is not pressed flat. The sampling configuration using the hydraulic cutter was practiced with an inactive stainless-steel pipe and approximately 5 mm height samples were cut. A 5 mm sample instead of a minimum of 2 to 3 cm sample was a major improvement as a smaller sample size resulted in a lower dose rate. Additionally, the need for additional sub-sampling was eliminated as the samples were sub-sampled directly after sampling using the hydraulic cutter. Two considerations were needed (1) the decontamination of the rust inside the tube would be needed prior to characterisation, and (2) the material was very hard. Therefore, the 5 mm samples were held with the pincers and then vertically cut twice resulting in four sub-subsamples out of which two sub-samples had exposed inner surfaces and two sub-samples were U-shaped without exposure to the inner surface (Fig. 2). Decontamination of the inner surface rust from the first two sub-samples was easy and characterisation studies were promptly initiated with minimal effort i.e., one day of work compared to several days of work.
Elemental analyses results
The sub-sampling requirements focused on the preparation of purpose-fit samples for ICP-MS, ICP-OES, ETM and DTM analyses. However, the requirements for GD-OES and gas analyser were not appropriately considered in the planning phase. GD-OES measurements required a flat sampling surface and therefore, polishing of the sample was carried out using silicon carbide sandpaper (500 grit) and a grinding machine. The polishing also removed impurities and oxide layers from the surface. The initial sample length was too long for the adapter and approximately 5 mm was cut from one end of the sample using a mechanical cutter. Less work would have been needed if the height of the sample would have been initially optimal (maximum of 20 mm) and EDM could have been used to form a flat surface. Secondly, the nitrogen measurements required several approximately 5 × 5 × 5 mm pieces, which were washed in acetone prior to analysis. In theory, the semi-destructive GD-OES analysis could have been carried out first followed by the nitrogen analysis after cutting the pipe into small pieces. However, in practice, the availabilities of the equipment demanded a second sampling campaign in order to accommodate both analyses.
Dissolution of inactive sub-sample was tested using aqua regia on a hot plate in preparation of ICP-OES samples. Small insoluble residue was removed with filtering as it did not dissolve completely in this media. The black residue was considered to contain carbon and consequently not affect the bulk elemental analysis. The ICP-OES analysed elements were main elements of austenitic steel (i.e., Si, P, S, Cr, Mn, Fe, Ni) and other interesting elements (i.e., Mn, Co, Mg, Cu). The low-activity samples for minor element analysis using ICP-MS were dissolved using a Milestone UltraWave microwave oven and conc. HNO3 and HCl in order to obtain complete dissolution. The measured elements were Nb, Mo, Ag, W, Zn, Ga, and Se. The measured elements and their mass fractions are summarised in Table 1. The combined ICP-MS and ICP-OES results converted to mass-percentages show that the studied steel is most likely an austenitic stainless steel due to its Cr and Ni contents. Additionally, the Mo results show that the stainless steel is not acid resistant (i.e., mass% was 0.25). The mass percentages determined by two independent methods (i.e., destructive ICP-MS and ICP-OES versus semi-destructive GD-OES) correspond quite well for the most abundant elements in this stainless steel, which are Fe, Cr, Ni, and Mn. However, there are large differences between the mass percentage values obtained by DA and semi-DA analysis for lower abundance elements. Low mass fractions of those elements in the stainless steel increase the measurement uncertainties of both methods.
The results of the gamma spectrometric measurement for the low-activity sample B are presented in Table 3. As presented, it was possible to quantify the 60Co activity by using gamma spectrometry in this sample even though the handheld devices were not able to detect the dose rate above the background. The measurement of sample B in the hot cell (with Detector 1) was not successful because of a high background level due to the presence of a radioactive waste bin inside the hot cell. The result of the activity of the highly active sample C measured inside the hot cell is an estimate as there are significant uncertainties in the efficiency calibration parameters e.g., the distance and the angle from the detector are not exact. The comparison of the activity of sample C with the sub-samples C1, C2, and C3 shows that averaged sub-sample result (6.21 × 105 Bq g−1) is 1.6 times higher than the value for Sample C (3.91 × 105 Bq g−1). The correlation between the Detector 1 and 2 measurement results can be estimated as relatively good considering that the Detector 1 measurement configuration has been designed for the measurement of larger sample sizes when the effect of the measurement parameters (i.e., distance and angle) is decreased. However, the relative standard deviation (RSD) between the Sub-samples C1–3 is 10.6%, which is approximately 10 times higher than expected. For example, the RSDs of the measured key radionuclide (i.e., 60Co, 152Eu, 137Cs) activity concentrations in homogeneity studies of previous intercomparison exercises were 1–2% [2, 4, 9,10,11,12]. Therefore, it was suggested that there are micro scale heterogeneities in small samples and also the neutron flux is heterogeneous, which both can cause variations in local specific activities. This phenomenon is further discussed in the intercomparison exercise results.
Each partner received approximately 0.1 g samples (named C1–C3), which consisted of several small chips (see Fig. 2 picture d) and they carried out the ETM and DTM analyses based on their internal procedures. The overall procedures of each partner are discussed separately as the developments of the analysis are described. The information given to all partners prior to analysis was the chemical composition, the preliminary activation calculation results and the average activity concentration of 60Co for the three sub-samples, pointing out possible microscale heterogeneity as the RSD of the sent samples was relatively large (Fig. 4).
Sample C1 analyses were first carried out for individual pieces of the highly activated steel (sub-samples C1.1 and C1.2) according to Leskinen et al. [3]. Selection of the sample masses was influenced by the amounts of stable Fe and Ni in the studied material as (i) the Fe capacity of 10 g of AG anion exchange resin is 4 mg and (ii) the Ni capacity of 0.5 g of Ni-resin is 2 mg. As the main bulk of the studied material was Fe, it was considered to be the limiting factor in the sample masses. Additionally, the representativeness and possible heterogeneity of the sample masses were considered. Calculations taking into account all these factors were carried out to determine sample masses, which would not require significantly larger amounts of AG resin to carry out the analysis. Consequently, the sub-sample masses were C1.1 (15.8 mg) and C1.2 (8 mg) (Table 4). Sub-sample C1.1 and C1.2 were acid digested prior to gamma analyses. The separation of Fe and Ni required several AG separations (using 10 g of resin) but only one purification step for the further purification of the Ni (using 0.5 g of Ni-resin). The Ni fractions after the Ni-resin purification were studied for 60Co contamination using HPGe gamma spectrometry and no further purifications were performed as no activity above the limit of detection was measured. The yield determination was carried out using ICP-OES and the activity was measured on a Hidex SL300 liquid scintillation counter. The sub-samples C1.1 and C1.2 show approximately 20% difference in 55Fe and 63Ni results and approximately 10% difference in 60Co results (see Figs. 5, 6 and 7), which were attributed to possible heterogeneity of the material. Therefore, another series of analyses were carried out on sub-sample C1.3 with a sample size significantly larger compared to sub-samples C1.1 and 1.2 (i.e., 61.1 mg). The sub-sample C1.3 was acid digested and measured for 60Co. Aliquots, which contained approximately 4 mg of Fe each, were taken for 55Fe and 63Ni analyses (sub-samples C1.3a, C1.3b and C1.3c). The measured activities for 55Fe and 63Ni for the sub samples C1.3 showed a lower RSDs value (1% and 2%, respectively) than the one obtained for sub-samples C1.1 and C1.2 (15% and 13%, respectively), confirming the micro-scale heterogeneity of sample C. The heterogeneity may originate from the heterogeneity of the metal or from variations in the induced activities due to gradients in the flux or energy distribution of the neutrons.
Sample C2 analysis approach included two main features. The first one was the selection of the suitable mass for the subsample. The subsample should contain adequately both stable and radioactive Fe and Ni isotopes regarding successful radiochemical separation, yield determination and radioactivity measurements, but still Fe and Ni content of the subsample should not exceed the binding capacity of chromatographic resin columns. Taking into account the binding capacities of the TRU and Ni resins for Fe and Ni, respectively, and the estimated mass fractions of Fe and Ni in the examined stainless steel, it was decided to use sub-sample masses below 10 mg for sample C2. Secondly, since the information from preliminary gamma measurements of this stainless steel indicated some heterogeneity of the material, it was considered necessary to combine and digest small subsamples for having a more homogeneous sample material prior to radiochemical separation of 55Fe and 63Ni.
Sub-samples C2.1, C2.2 and C2.3 were first measured by gamma spectrometry before these steel fragments were combined and digested in aqua regia. The solution was then divided into three parts, i.e., sub-samples C2.123a, C2.123b and C2.123c. Sample pooling prior to dissolution and later division into subsamples again was performed to obtain more homogenous samples to perform the radiochemical separations, compared to the option of digesting the individual solid sub-samples C2.1, C2.2 and C2.3 separately. Sub-samples C2.5a and C2.5b were prepared similarly so that after the digestion of sub-sample C2.5 was split into two sub-samples. The separation method for 55Fe and 63Ni used on sample C2 was modified from [4, 27,28,29], starting from iron hydroxide precipitation of Fe and Ni and continuing with column separations. Due to the small sample size and the resulting low amount of stable Fe and Ni, it was possible to separate Fe and Ni from each other and other interfering radionuclides using a single TRU resin column with 0.7 g of resin. The Ni-fraction was further purified with 0.7 g of Ni resin to remove the Co. Ni resin column separation had to be repeated once or twice, depending on the residual activity of 60Co in the purified Ni fraction. The activity concentration of 60Co in the purified Fe and Ni fractions was monitored with gamma spectrometric measurements. It was found out, that the 60Co activity in the Ni fractions was first decreased by approximately 90% after the second purification round, while the 60Co activity was decreased by approximately 70% after the third purification round. The activity of 60Co was determined with HPGe detector and of 55Fe and 60Ni by TriCarb 2910 TR LSC. Yield determination for sample C2 sub-samples was performed by ICP-MS by a subcontractor.
As the calculated activity concentration of the material was in the order of several thousand becquerels per gram, it was decided to handle only 10 mg of sample C3 per batch of analysis. The chemical composition of the steel (given as initial data) confirmed that 10 mg was in agreement with the maximum amount (0.1 g of sample) that could be analyzed with the in-house method adapted from [30]. The sub-samples C3.1, C3.2 and C3.3 were dissolved overnight using aqua regia prior to gamma measurement using HPGe detector. An aliquot of sub-sample C3.1 was taken before further analysis, while the entire sub-samples C3.2 and C3.3 were treated. The method for the separation of 55Fe and 63Ni started with the addition of a carrier before an iron oxide precipitation. AG1 × 4 anion exchange resin was used to isolate the Fe fraction while the Ni fraction was further purified using Ni-resin. The 60Co, 55Fe and 60Ni activities were measured on a Quantulus liquid scintillation counter. The yield determination for sample C3 sub-samples was performed by ICP-OES. The 55Fe, 63Ni and 60Co results for sub-samples C3.1, C3.2 and C3.3 show similar variance to sample 1 which seems to confirm the hypothesis that the sample is heterogenous in micro-scale.
For samples C1-3, yields of 55Fe and 63Ni were determined by adding stable Fe and Ni carriers (if needed) at the beginning of the analysis and by measuring the mass fractions of the stable Fe and Ni from the acid digested solutions (i.e., verification of original Fe and Ni contents) and the final purified fractions by ICP-OES or ICP-MS. The high uncertainties in the Fe and Ni yields of sample C2 are due to high relative uncertainties of Fe and Ni mass fractions announced by the subcontracting ICP-MS laboratory, being 20% for Fe and 25% for Ni, respectively. For samples C2 and C3, the 55Fe yields are higher than the 63Ni, one which can be explained by the fact that a second purification process was used for all the methods.
The standard and robust statistics in Table 5 were calculated with selected sub-sample results (maximum of 3 results/radionuclide/sample series) as the same weight by each participant was desired. The selected 55Fe results were the first three for the sample C1 sub-sample and all sample C3 sub-sample results. As discussed later, re-calculations were carried out for the sample C2 sub-sample results and only two results were well aligned with the other results (Fig. 5). The first three 63Ni results were selected from the sample C1, C2 and C3 sub-samples as no clear outliers were seen (Fig. 6). The selected 60Co results were only those which had been carried out for acid digested samples as differences between measurements carried out as point sources and in acid digested samples were observed and the gamma analysis results from acid digested samples were considered more reliable (discussed later).
The 55Fe results with the assigned value with k = 2 uncertainties are shown in Fig. 5. Sample C1 and C3 sub-samples are well aligned whereas the initial sample C2 sub-samples (blue cross) are consistently above the general trend. The most likely reason for the deviation was concluded to arise from the efficiency calibration of the TriCarb liquid scintillation counter, which had been carried out using an old 55Fe standard solution, and also from using a measurement protocol designed for 3H, as that was the closest option to 55Fe, among the existing protocol options. Even though 3H and 55Fe are low energy emitters, 55Fe decays with electron capture and thus it is extremely easily influenced by even small changes in quenching [31]. Therefore, the sample C2 sub-samples were re-measured using Hidex 300 SL LSC using TDCR and CoreF function. The re-calculated sample C2 sub-samples results (blue dot) in Fig. 5 show that the first three 55Fe results remain above the general trend whereas the final two results are well aligned with the general trend. First two analyses of both samples C1 and C3 sub-samples were carried out for individual pieces of steel (black dot) whereas the rest of the analyses were carried out for aliquots (blue dot). No clear bias between the aforementioned ways of analyses can be concluded even though the relative standard deviation (RSD) of aliquot samples C1.3a, C1.3b, and C1.3c is only 1.1% whereas the RSD of all sample C1 sub-sample analyses is 7.8%. The assessment of the 2k uncertainties concluded that the sample C3 sub-samples results were most likely underestimated due to an underestimation of the yield uncertainty and uncertainties for 55Fe specific activity of sample C2 sub-samples were large due to fore mentioned high uncertainty in yield.
The final 63Ni results and the assigned value with k = 2 uncertainties in Fig. 6 show that all the results are well aligned. However, similarly to the 55Fe results, the uncertainties of sample C3 sub-samples are most likely underestimated and the uncertainties of sample C2 sub-samples are significantly large. Additionally, no clear dependence between the analysis of individual pieces of steel and aliquots is seen.
The 60Co results with the assigned value with k = 2 uncertainties in Fig. 7 show that all sample C1 and C3 sub-samples have been measured after acid digestion whereas initially submitted sub-samples C2.1, C2.2, C2.3, and C2.5 results were measured as individual pieces (point source). These results were discussed, and it was concluded that the measurements in acid-digested samples with efficiency calibration for fixed volume seemed to produce more comparable results compared to the results obtained by solid sample measurement and point source type efficiency calibration. The hypothesis was studied by measurement of sub-samples C2.4.1 and C2.4.2 both as point sources and after acid digestion. The results show a clear trend that the point source calibrations give approximately 10% higher activity values than the volume calibrations. However, comparisons of Table 2; Fig. 6 60Co results show that all the initially measured gamma activities of complete sub-samples C1–C3, which were measured using point source geometry calibrations, correlate with the assigned value and expanded uncertainty (2k). A reason for the good correlation is most likely attributed to the averaging phenomenon of several fragments of samples C1–C3 with larger masses in contrast to individual fragments with smaller masses and the effect of measuring at relatively large distance from the detector.

Final 55Fe specific activities and the assigned value with expanded uncertainties (k = 2) in the trilateral intercomparison exercise. Red triangles indicate analyses in which individual pieces of sample were analysed and blue dots indicate analyses in which aliquots were analysed. Blue crosses were initial results calculated with uncorrect efficiency curve

Final 63Ni specific activities results with expanded uncertainties (k = 2) in the trilateral intercomparison exercise. Red triangles indicate analyses in which individual pieces of sample were analysed and blue dots indicate analyses in which aliquots were analysed

Final 60Co specific activities results with expanded uncertainties (k = 2) in the trilateral intercomparison exercise. Black dots indicate analyses in which individual pieces of sample were analysed and red diamonds indicate analyses carried out for acid digested samples
Even though the assigned values were calculated using selected results (see Table 5), the z scores were calculated for all the submitted results (Table 6). The 55Fe z score results show that all sample C1 and C3 sub-sample results are within the acceptable range (|z| ≤ 2.0). Three out of five sample C2 sub-sample results are in unacceptable range (|z| ≥ 3.0) whereas the remaining 2 results are in acceptable range. The 63Ni z score results show that all sample C1, C2 and C3 sub-sample results are in acceptable range. Sample C1 and C3 sub-sample 60Co z scores are all in acceptable range whereas one out of eight sample C2 sub-sample analysis result is in unacceptable range and two is given a warning signal (2.0 < |z| < 3.0).
The 55Fe/60Co and 63Ni/60Co scaling factors are shown in Table 7. The sample specific averaged 55Fe/60Co scaling factors for sub-samples C1, C2 and C3 are 1.2, 1,7 and 1.1, respectively whereas the corresponding 63Ni/60Co scaling factors are 1.4, 1.2 and 1.3. The averaged 55Fe/60Co scaling factor for all results is 1.3 with 21% RSD. The corresponding 63Ni/60Co averaged scaling factor is 1.3 with 9% RSD. Sub-sample C2 55Fe/60Co scaling factors are higher than the corresponding values of sub-samples C1 and C3 causing the relatively large RSD (i.e., 21%) As previously discussed, the activity concentration of 60Co in these samples obtained by point source efficiency calibration was about 10% higher than the general activity concentration level of other samples, determined by volume-type efficiency calibration. However, a similar phenomenon is not seen for the averaged 63Ni/60Co scaling factor (i.e., 9% RSD). The averaged 55Fe/60Co and 63Ni/60Co scaling factors calculated using only sub-sample C1 and C3 results are 1.1 (5% RDS) and 1.4 (8% RSD), respectively.
Activation calculation results and comparison with measurement results
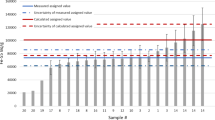
The specific activities in the activated steel pipe between 0 and 12 cm with 1 cm increments calculated with MCNP and ORIGEN-S are shown in Figs. 7, 8 and 9 together with measured assigned values. Uncertainties were calculated using the law of error propagation in multiplication. The uncertainties arise from the uncertainties in the exact original material composition, neutron dose, reaction cross sections and decay time. ORIGEN-S uses ENDF/B-VI cross Sections. 10% uncertainty was estimated for cross sections. Element-wise uncertainties in material compositions were estimated from measurement uncertainties listed in Table 1. ICP-OES and ICP-MS results were utilised in all cases except for N and C. 10% uncertainty was also assumed for total operating hours. Therefore, the calculated overall uncertainty for 60Co was 18.9%, for 63Ni 16.6% and for 55Fe 16.4%.
Neutron flux contains much higher uncertainties since it is impossible to calculate the flux for a very small volume representing the sampling point. Moreover, the flux above the core grid does not decrease smoothly as a function of distance since the surrounding materials can be considered as a disk source (not as a point source) and epithermal and fast neutron thermalise in the surrounding water. In years 1987–2015 there have also been several fuel loading pattern changes, which affect the fluxes slightly. Therefore, activity concentrations were calculated for distances 0–10 cm from the surface of the core.
Comparison of the calculated and measured (i.e., assigned values) specific acitivties of 55Fe, 63Ni and 60Co in Figs. 7, 8 and 9 show that in all cases the calculated and measured results align between 6.5 and 8 cm distance. As discussed before, approximately first 5 cm of the instrumental pipe was removed with the fuel element in 2020 campaign and the sub-sampling was carried out without knowing which end of the sample C was attached to the removed fuel element (i.e., highest activity). However, the sub-sampling distance should be approximately either 5–6 cm (i.e., highest activity end of sample C) or 8–9 cm (i.e., lowest activity end of sample C). Therefore, it can be concluded that the sub-sampling was carried out from the lowest activity end of sample C. Additionally, the results show excellent correlation between the activation calculation and experimental results.

Calculated and measured (assigned value) 55Fe activity concentration results with expanded uncertainties (k = 2). The cross-section of the calculated and measured activities are shown as an excerpt

Calculated and measured (assigned value) 63Ni activity concentration results with expanded uncertainties (k = 2). The cross-section of the calculated and measured activities are shown as an excerpt

Calculated and measured (assigned value) 60Co activity concentration results with expanded uncertainties (k = 2). The cross-section of the calculated and measured activities are shown as an excerpt
Conclusions
Careful planning of the complete sampling campaign was essential with such a high activity sample material as the stainless steel of this study. Both radiation safety and radioanalytical requirements need to be considered and compromises must be often made while fulfilling these partly conflicting requirements. The most important questions before the sampling were the quality of the samples, e.g., activity level, adequate mass for analyses regarding instrumental detection limits and absorption capacities of ion exchange and chromatographic resins, estimated specific activities of both radionuclides and stable elements of interest, and homogeneity of the samples. It was found that the best strategy to avoid unnecessary intermediate steps in sample dissolution and overcome the microscale heterogeneity, which is problematic with small sample sizes, was to first dissolve a larger sample mass and then to divide it into aliquots for the analyses. The micro-scale heterogeneity was already seen in the homogeneity studies using gamma spectrometry when the RSDs were higher than expected.
The intercomparison exercise results were overall well aligned. It can be concluded that in this study, there was a clear benefit in measurement of the gamma activities with volume-type efficiency calibration in contrast to point source efficiency calibration. Additionally, the experimentally determined assigned values and activation calculation results exhibited excellent correlation.
References
Kutz M (2002) Handbook of materials selection. Wiley, New York
Leskinen A, Tanhua-Tyrkkö M, Kekki T, Salminen-Paatero S, Zhang W, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Stordal S, Fichet P, Gautier C (2020) Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-429 report, ISBN 978-87-7893-519-9, Roskilde, Denmark
Leskinen A, Salminen-Paatero S, Räty A, Tanhua-Tyrkkö M, Iso-Markku T, Puukko E (2020) Determination of 14 C, 55Fe, 63Ni and gamma emitters in activated RPV steel samples - a comparison between calculations and experimental analysis. J Radioanal Nucl Chem 323:399–413
Leskinen A, Salminen-Paatero S, Gautier C, Räty A, Tanhua-Tyrkkö M, Fichet P, Kekki T, Zhang W, Bubendorff J, Laporte E, Lambrot G, Brennetot R (2020) Intercomparison exercise on difficult to measure radionuclides in activated steel - statistical analysis of radioanalytical results and activation calculations. J Radioanal Nucl Chem 324:1303–1316
Corcho-Alvarado JA, Sahli H, Röllin S, von Gunten C, Gosteli R, Ossola J, Stauffer M (2020) Validation of a radiochemical method for the determination of 55Fe and 63Ni in water and steel samples from decommissioning activities. J Radioanal Nucl 326:4555–4463
Houk RS (1986) Mass spectrometry of ICPs. Anal Chem 58:97A–105A
Luong ET, Houk RS (2003) Determination of carbon isotope ratios in amino acids, proteins, and oligosaccharides by inductively coupled plasma-mass spectrometry. J Am Soc Mass Spectrom 14:295–301
Hou X, Togneri L, Olsson M, Englund S, Gottfridsson O, Forsström M, Hironen H (2016) Standardization of radioanalytical methods for determination of 63Ni and 55Fe in waste and environmental samples. NKS-356 report ISBN 978-87-7893-440-6, Roskilde, Denmark
Leskinen A, Tanhua-Tyrkkö M, Salminen-Paatero S, Laurila J, Kurhela K, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Laporte E, Giuliani M, Bubendorff J, Fichet P (2021) DTM-Decom II - Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-441, ISBN 978-87-7893-533-5 Roskilde, Denmark
Leskinen A, Gautier C, Räty A, Fichet P, Kekki T, Laporte E, Giuliani M, Bubendorff J, Laurila J, Kurhela K, Fichet P, Salminen-Paatero S (2021) Intercomparison exercise on difficult to measure radionuclides in activated concrete - statistical analysis and comparison with activation calculations. J Radioanal Nucl Chem 329:945–958
Leskinen A, Dorval E, Salminen-Paatero S, Hou X, Jerome S, Jensen KA, Skipperud L, Vasara L, Rautio S, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Baudat E, Lambrot G, Giuliani M, Colin C, Laporte E, Bubenforff J, Brennetot R, Wu S-S, Ku H, Wei WC, Li YC, Luo QT (2022) DTM-Decom III - intercomparison exercise in analysis of DTM beta and gamma emitters in spent ion exchange resin. NKS-457, ISBN 978-87-7893-550-2, Roskilde, Denmark
Leskinen A, Dorval E, Baudat E, Gautier C, Stordal S, Salminen-Paatero S (2023) Intercomparison exercise on difficult to measure radionuclides in spent ion exchange resin. J Radioanal Nucl Chem 332:77–94
Leskinen A, Lavonen T, Dorval E, Salminen-Paatero S, Meriläinen V, Hou X, Jerome S, Jensen KA, Skipperud L, Rawcliffe J, Bourgeaux-Goget M, Wendel C, Stordal S, Isdahl I, Gautier C, Taing Y, Colin C, Bubendorff J, Wu S-S, Ku YH, Li YC, Luo QT (2023) RESINA – intercomparison exercise on alpha radionuclide analysis in spent ion exchange resin. NKS-466, ISBN 978-87-7893-560-1, Roskilde, Denmark
Leskinen A, Hokkinen J, Kärkelä T, Kekki T (2022) Release of 3H and 14 C during sampling and speciation in activated concrete. J Radioanal Nucl Chem 331:859–865
Räty A, Lavonen T, Leskinen A, Likonen J, Postolache C, Fugaru V, Bubueanu G, Lungu C, Bucsa A (2019) Characterisation measurements of fluental and graphite in FiR1 TRIGA research reactor decommissioning waste. Nucl Eng Design 353:110198
Räty A, Kekki T, Tanhua-Tyrkkö M, Lavonen T, Myllykylä E (2018) Preliminary waste characterisation measurements in FiR1 TRIGA research decommissioning project. Nucl Tech 203(2):205–220
Arffman P, Lydman J, Hytönen N, Que Z, Lindqvist S (2022) BRUTE: evaluation of mechanical properties of true reactor pressure vessel material from barsebäck 2. ASME 2022 pressure vessels and piping conference, materials and fabrication, vol 4A PVP 2022–83819, ISBN 978-0-7918-8617-5
Hytönen N (2019) Effect of microstructure on brittle fracture initiation in a reactor pressure vessel weld metal. Master of science thesis, Tampere University, Finland
Dulski TR (1996) A manual for the chemical analysis of metals. ASTM manual series: MNL 25. American Society for Testing and Materials. https://doi.org/10.1520/MNL25-EB
Scheffler GL, Brooks AJ, Yao Z, Daymond MR, Pozebon D, Beauchemin D (2016) Direct determination of trace elements in austenitic stainless steel samples by ETV-ICPOES. J Anal at Spectrom 31:2434–2440
Analysis of Stainless Steel by Dual View Inductively Coupled Plasma Spectrometry (2023) Teledyne leeman labs. Application note – AN1505, 28 May 2015. http://www.teledyneleemanlabs.com/resource/Application%20Notes/AN1505_Analysis%20of%20Stainless%20Steel%20by%20Dual%20View%20ICP-OES.pdf. Accessed 12
Maxwell SL, Culligan B, Hutchinson JB, Sudowe R, McAlister DR (2017) Rapid method to determine plutonium isotopes in steel samples. J Radioanal Nucl Chem 314:1103–1111
International Standard ISO 13528:2015(E), statistical methods for use in proficiency testing by interlaboratory comparison. ISO, Geneva, Switzerland
X-5 Monte Carlo Team (2003) MCNP – a general monte carlo nparticletransport code, version 5, Los Alamos National Laboratory, LA-UR- 03-1987
Herman IC, Westfall OW (2011) RM ORIGEN-S: a scale system moduleto calculate fuel depletion, actinide transmutation, fission product buildup and decay, and associated radiation source terms, version 6.1, 2011. Oak Ridge National Laboratory, ORNL/TM-2005/39
Räty A (2020) Activity characterisation studies in FiR1 TRIGA research reactor decommissioning project, doctoral thesis, Helsinki University, Finland
Triskem International (2014) Iron-55 in water. Analytical procedure. Method number FEW01. Revision 1.1, 1st May 2014
Eriksson S, Vesterlund A, Olsson M, Ramebäck H (2013) Reducing measurement uncertainty in 63Ni measurements in reactor coolant water with high 60Co activities. J Radioanal Nucl Chem 296:775–779
Triskem International (2014) Nickel 63/59 in water. Analytical procedure. Method number NIW01. Revision 1.3, 1st May 2014
Hou X, Østergaard LF, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste samples using radiochemical separation and liquid scintillation counting. Anal Chim Acta 535(1–2):297–307
Kojima S, Furukawa M (1985) Liquid scintillation counting of 55Fe applied to air-filter samples. Radioisotop 34:72–77
Acknowledgements
The authors would like to thank the Finnish Research Programme on Nuclear Waste Management KYT 2022 for funding. FiR1 decommissioning project colleagues for collaboration and provision of studied material.
Funding
Open Access funding provided by Technical Research Centre of Finland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing financial interests or personal relationships with people or organisations that could have inappropriately influenced the work presented in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leskinen, A., Salminen-Paatero, S., Leporanta, J. et al. Sampling, characterization, method validation, and lessons learned in analysis of highly activated stainless steel from reactor decommissioning. J Radioanal Nucl Chem 333, 145–163 (2024). https://doi.org/10.1007/s10967-023-09182-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-023-09182-y