
Abstract
A new rapid method has been developed for the determination of Th, Pu, Np, U, Am and Cm isotopes in water samples of about 1 L. Actinides are pre-concentrated by co-precipitation with Ca phosphate, sequentially separated on stacked TEVA and TK221 cartridges and measured by alpha spectrometry. The TK221 extraction chromatographic resin contains i.e. CMPO and DGA extractants. It has been characterized by measuring the weight distribution ratios (Dw) of actinides which are higher than 1000 for all actinides in 3 M HNO3. The method has been optimized, applied for the analysis of tap and seawater samples and validated by participating in an IAEA proficiency test. Chemical recoveries for all actinides are better than 50%. The method can be performed within one day.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A great variety of radiochemical methods have been developed for the simultaneous determination of all actinides including the man-made nuclides of Np, Pu, Am, Cm and the nuclides of natural origin such as Th and U. Methods of high sensitivity are of special interest for monitoring the environment and rapid methods are essential for detecting contamination or leaks in emergency situations. Methods that can meet both the demands of moderate sensitivity and speed are very useful in controlling nuclear technology, managing nuclear incidents. After the accident at Fukushima NPP in 2011 an urgent need for accurate and fast determination of actinides especially in water and sea water samples arose what initiated a development of rapid analytical techniques.
Conventional procedures for analysis of actinides were based on ion exchange, liquid–liquid extraction and a series of precipitations, but they were rarely selective for all actinides and therefore long and tedious. In course of the years specific extractants have been developed for the nuclear industry, but later some of them have been introduced into the analytical practice often as extraction chromatographic (EC) resins. Horwitz et al. developed and characterized the carbamoylmethyl phosphine oxide (CMPO) and the tributyl phosphate (TBP) coated resin, later called TRU resin [1] in 1986, the liquid anion exchanger Aliquat® 336 coated resin that got the name TEVA resin [2] in 1992, and in the same year the dipentyl(pentyl) phosphonate coated resin, later called UTEVA resin [3]. These resins were able to retain selectively certain actinides from acidic solutions. Increasing the acidity the retention of actinides were improved, either as ion association complexes or as anionic species. By the middle of the nineties a set of selective actinide separations could be realized: tetravalent actinides such as Th, Pu(IV) and Np(IV) could be retained on TEVA, tetra- and hexavalent actinides including U(VI) could be extracted by UTEVA. Further to all of these actinides, the trivalent ones like Am and Cm–although with smaller weight distribution ratios (Dw)–could be retained on TRU resin.
From that time on a busy period of method development for actinides determination in various sample types started and the new methods began to replace the conventional ones. Combinations of EC resins provided high decontamination factors (DF) for given actinides against other ones. In these combined systems TRU resin was always included due to its unique feature to retain trivalent actinides.
In the TEVA-UTEVA-TRU resin combination first proposed by Horwitz in 1995, Th and Pu were retained on TEVA, U on UTEVA and Am, Pu(III) on TRU resin [4]. The use of three or more columns made the procedure expensive, therefore two-column approaches were followed by many authors.
A successful method for determination of all actinides was based on UTEVA-TRU resins where U and Th were retained from 3 M HNO3 on UTEVA while Am and the reduced form of Pu(III) were retained on TRU resin followed by their sequential elution [5,6,7]. In the TEVA-TRU separations tetravalent actinides (Th, Pu(IV), occasionally Np(IV)) were retained on TEVA resin, while trivalent (Am, Cm) and hexavalent actinides (U(VI)) were separated on TRU resin. This combined method for Pu and Am separation was used by Ayranov et al. in 2005 [8] for determination of Pu and Am nuclides in soil samples. The Dw values of actinides on TEVA from nitric acid are significantly higher than on UTEVA thus allowing a stronger retention of actinides. The weak point of these separations is the use of TRU resin for the retention of trivalent actinides because the maximum Dw values are about 100 (from 1 to 2 M HNO3) and the presence of Fe even after reduction to Fe(II) will further reduce the retention. Additionally, lanthanides are co-extracted with trivalent actinides therefore lanthanide–Am, Cm separation is often required most likely using a TEVA or anion exchange resin column.
The solution to eliminate this failure arrived with the introduction of a new material, the diglycolamide coated DGA resins in the analytical practice by Horwitz et al. in 2005 [9]. The unique feature of DGA resins is the increasing distribution coefficient of trivalent actinides with increasing HNO3 or HCl concentration ending up at a value of > 1000 at 3–4 M acidity. This allows the strong retention of Am, Cm on DGA and their elution with dilute acids. Furthermore, there is a certain selectivity for trivalent actinides against lanthanides (especially light lanthanides). DGA resins are available either with normal (DGA-N) or branched (DGA-B) alkyl chains in the extractant molecule. For environmental applications, mainly DGA-N resin is used, for it retains actinides (especially Am) better than DGA-B.
The methods for actinide separations started to include DGA resin in the protocol. Maxwell et al. proposed first to add DGA to the TEVA-TRU combined procedure for processing water samples in 2006 [10]. In the procedure Th, Np(IV) and Pu(IV) are retained from 3 M HNO3 on TEVA and stripped as Th, and Np-Pu common fractions. Americium (partially) and U are retained on stacked TRU resin. Then Am is transferred from TRU to DGA resin with 3–4 M HCl rinse. Finally, U is stripped from TRU and Am from DGA resin. Maxwell et al. adopted this separation for various matrices such as water, seawater [11], urine [12], soil, sediment [13], concrete, bricks [14], asphalt [15] and biological material [16]. He also proposed the use of calcium phosphate for pre-concentration of actinides in water, and the use of sulfamic acid and ascorbic acid followed by the addition of NaNO2 to adjust the oxidation states of Np and Pu together to tetravalent which is not an easy process. In these methods he added aluminum nitrate to the load to complex phosphates and form salting-out agent. In case of solid samples he recommended fusion with NaOH to dissolve different matrices rapidly. His method for water analysis became the basis of the recommended procedures of Eichrom (ACW16 [17] and ACW17 [18]) and Triskem (TKI-AC-01), the two companies providing EC resins for actinide analysis. Since the introduction and commercial availability of DGA resin many new combined procedures have been developed and reported world-wide. We made a summary of the methods published in the last 20 years that aim the determination of all actinides in water samples, results are presented in Table 1.
Attempts have also been made to analyze all actinides on a single EC column, i.e. on TRU or DGA resins. In our laboratory we developed a rapid combined procedure for separation of Am, Pu, Th and U (later Np was also involved) from soil samples using TRU resin [28]. This method perfectly fit the purposes of rapid determination of all actinides by alpha spectrometry, but the column could not be loaded with bigger amounts of sample due to the limited retention of Am. After the appearance of DGA resin on the market we developed a new combined procedure that could handle bigger amounts (up to 5 g) of soil [29]. The methods based on the use of a single resin are definitely cheaper, but DFs for given actinides are often smaller than in case of the multi-column methods which can be regarded as repeated separations on the consecutive columns. Furthermore, in multi-column systems cross-contaminations of the actinides are easier to avoid, rinsing the columns between the consecutive stripping of individual actinides is usually not needed, altogether less fractions (strip and wash) are collected. For the novel rapid method to determine actinides in water therefore our preference was the multi-column approach, we studied the TEVA-TRU-DGA combined procedure of Maxwell et al. [10].
Although the obtained analytical results and the reported method performances (yields, DF values) in the TEVA-TRU-DGA combined procedures of Maxwell et al. are excellent, the use of three different resins and their repeated couplings make the procedure somewhat requiring. We propose an easy-to-perform improved procedure for the rapid determination of all actinides in water samples. The extractants of TRU and DGA resins were combined into a new resin of Triskem, the TK221 resin, which is i.e. available in 2 mL cartridges. In the novel method stacked TEVA and TK221 resin cartridges are used. Th, Pu(IV) and Np(IV) are retained together and individually eluted from the separated TEVA, while Am(Cm) and U are retained and sequentially eluted from TK221. The resin was characterized by batch uptake experiments, retention and elution conditions were optimized. The method was validated by participating in a proficiency test organized by International Atomic Energy Agency (IAEA).
Experimental
Materials and equipment
Chromatographic resins: 2 mL cartridges of TEVA and TK221 resins were purchased from Triskem International SAS, France. TEVA resin contains Aliquat® 336 (N-methyl-N,N,N-trioctylammonium chloride) extractant coated on Amberchrom™ CG71 inert support. TK221 resin is regarded as a combination of TRU and DGA-N resins. The active component of TRU resin, octyl(phenyl)-N,N-diisobutyl-carbamoylmethyl phosphine oxide and that of DGA-N resin, N,N,N’,N’-tetra(1-octyl)-diglycolamide as a mixture of extractants are coated on PS-DVB inert support. 2 mL resin cartridges were used.
Reagents and materials: All reagents used (HNO3, Ca(NO3)2·4H2O, (NH4)2HPO4, Al(NO3)3·9H2O, Fe(NO3)3·9H2O, KSCN, sulfamic acid (NH2HSO4), ascorbic acid, NaNO2, HCl, HF, Mohr’s salt, TiCl3, NH3, ammonium bioxalate, neodymium oxide) were of analytical grade. Solutions were prepared with distilled water. Gold Star LS cocktail from Meridian, U.K. was used for LS measurements. Calcium phosphate precipitates were filtered through 0.45 µm pore size cellulose nitrate membrane. For alpha source preparation, cellulose nitrate membranes with 0.1 µm pore size were used.
Radioactive tracers: Carrier-free tracers of 239Pu, 230Th, 241Am used for test experiments were purchased from CERCA-LEA, France, 233U and 237Np tracers were purchased from New Brunswick Laboratory, U.S. DoE and AEA, U.K., respectively. Tracers of 242Pu, 243Am, 232U used for spiking water samples were purchased from NIST, U.S.
Samples
For testing the method tap and seawater samples were taken from our campus in Budapest and the Brighton seashore, UK. Seawater samples were filtered through 0.45 μm pore size membrane before use. Calcium content of the raw samples was measured by AAS. The Ca content of the tap water and the sea water was about 80 and 440 mg L−1, respectively.
For validation of the method water samples of the IAEA-TEL-2021-03 world-wide open proficiency test were used.
Methods
I. Determination of the distribution ratios
Batch uptake experiments were performed with 239Pu, 230Th, 241Am and 233U tracers. Nitric acid solutions of various concentrations were spiked with one of the tracers, the activity concentration of the solutions was in the range of 350–1200 Bq mL−1. When Pu was tested NaNO2 was added to each solution to obtain 0.01 M concentration. Then 1.5 mL of the acid solution was contacted with a known amount of resin (around 0.025 g) in a disposable plastic centrifuge tube. A mixing time of 1 h was employed. After equilibration the phases were separated by filtering the suspension through disposable membrane filters (0.45 μm pore size). The weight of the filtrate was measured and its volume was calculated from the measured density of the load solution. Therefore corrections due to filtration losses could be performed. To each set of samples (3 parallel runs) 1 standard and 1 blank sample were prepared. Liquid samples were mixed with 10 mL Gold Star LS cocktail. Weight distribution ratios (Dw) were calculated from Eq. (1):
where A0 and A are the aqueous phase activities before and after equilibration, respectively, w is the weight of the resin (g), V is the volume of the aqueous phase (mL). The k’ capacity factors were calculated from Dw values (mL g−1) with the conversion factor C: k’ = Dw × C where C was taken as approximately 0.56 g mL−1 for TK221 resin.
II. Optimization of the elution conditions
Individual radioactive tracers 239Pu, 230Th, 241Am and 233U of about 50 Bq were added to the test solutions modelling the composition of the load solutions originating from water samples after pre-concentration. The solutions contained the following components: 12 mL 3 M HNO3/1 M Al(NO3)3 spiked with 1 mL of the given tracer, 0.5 mL 1.5 M sulfamic acid, 1.5 mL 1 M ascorbic acid. The role of Al(NO3)3 is to provide high nitrate concentration to enhance the formation of actinide-nitrate complexes, while maintaining nitric acid concentration low enough to prevent the damage of the resins. Sulfamic acid and ascorbic acid are reducing agents which maintain the desired redox conditions. For given tests Ca and/or Fe carriers were also added as nitrate salts. Finally, 244 mg NaNO2 were added to the cold load, and the solution was left for half an hour. A given portion (1 mL) of the solution was taken as A0 reference for liquid scintillation counting (LSC), the rest was used as load solution.
Tests were performed with one resin cartridge or the stacked cartridges.
Elution tests were performed in vacuum box with the given resin cartridges and eluents at a flow rate of about 1–2 mL min−1. After the loading the columns were rinsed with 3 M HNO3. After separation of the columns, thorium was eluted with 9 M HCl. Plutonium and neptunium were eluted together with 0.1 M HCl/0.05 M HF/0.03 M TiCl3. If Pu and Np were eluted separately, Pu was eluted by on-column reduction to Pu(III) with 9 M HCl/0.03 M TiCl3 solution, and Np was eluted by 0.1 M HCl/0.1 M HF. Americium was with 0.25 M HCl, and U was eluted with 0.1 M NH4HC2O4. Eluent fractions were collected in LS glass vials. The liquid (except for the Ti containing solution) was evaporated to dryness, then taken up in 2.5 mL 0.3 M HNO3 and mixed with 15 mL Gold Star cocktail. The reference standard and the blank were prepared similarly. To the Ti containing solutions 25 μL 65% HNO3 was added, the solution lost the pink color immediately and 1.5 mL aliquot was directly mixed with the cocktail.
Measurements were performed with a TriCarb TR2800 LS counter. Measuring time was about 30 min. Measured count rates were related to the reference standard to obtain the % of eluted fraction of the given radionuclide.
III. Analysis of actinides in water samples
800 ml water sample was acidified with 4 mL 65% HNO3.
Pre-concentration with Ca phosphate
0.295 g Ca(NO3)2·4H2O (equivalent with 50 mg Ca) were added to the water sample. The samples were spiked with the following radiotracers: 230Th, 233U, 239Pu, 241Am (~ 0.5 Bq each) and 237Np (~ 0.1 Bq) and heated for 30 min. 3 mL 3.2 M diammonium hydrogen phosphate solution was added to the samples. The calcium phosphate was precipitated by adjusting the pH to 7 with 25% NH3 solution (approx. 8.5 mL). The samples were heated for 30 min and left to stand overnight. The calcium phosphate precipitate was filtered through a 0.45 µm pore size cellulose nitrate membrane. The precipitate was dissolved in 10 mL 65% HNO3 diluted with about 30 mL distilled water. The solution was evaporated to dryness. In order to destroy organic matter the evaporation was repeated with 65% HNO3 (2 mL for tap water sample, 5 mL for seawater sample).
Load preparation
12 mL 1 M Al(NO3)3/3 M HNO3 and 3 mL 0.25 M Fe(NO3)3/3 M HNO3 (equivalent with 14 mg Fe) solutions were added to the evaporation residue. The samples were heated in order to accelerate dissolution. 72 mg sulfamic acid and 264 mg ascorbic acid were added to the solution. After 15 min the solution was cooled to room temperature and 244 mg sodium nitrite was added and left to stand for at least 15 min.
Separation on TEVA/TK221 stacked columns
The stacked TEVA/TK221 cartridges were preconditioned with 10 mL 3 M HNO3 then the pre-prepared solution was loaded on. The required flow rate range is 0.5–1.0 mL min−1. The vessel of the load solution was rinsed with 3 mL 3 M HNO3.The rinse was loaded on the column as well. The columns were washed with 15 mL 3 M HNO3. The columns were separated and the actinides were sequentially eluted.
Elution of actinides from the TEVA column
The column was washed with 10 mL 3 M HNO3. Thorium was stripped with 15 mL 9 M HCl into a glass beaker. Pu was stripped into a Teflon beaker with 15 mL 9 M HCl/0.03 M TiCl3 solution. 1 mL 40% HF was added to the strip solution to prevent the precipitation of Pu. Np was stripped with 20 mL 0.1 M HCl/0.1 M HF solution. If Pu and Np separation was not required, they were stripped together with 20 mL 0.1 M HCl/0.05 M HF/0.03 M TiCl3 solution.
Elution of actinides from the TK221 column
The column was washed with 15 mL 4 M HCl/0.2 M HF. Am was stripped with 30 mL 0.25 M HCl. Uranium was stripped with 15 mL 0.1 M ammonium bioxalate solution.
Preparation of alpha sources
Alpha sources were prepared by micro-co-precipitation with NdF3. To each strip solution (directly or after a pretreatment) 50 µL Nd stock solution containing 1000 µg/1000 µl Nd3+ and 5 mL 40% HF were added and left to stand for 30 min. The micro-precipitate was filtered through a 0.1 µm pore size cellulose nitrate membrane.
Strip solutions were pretreated by the following procedures: The thorium strip solution was gently evaporated to near dryness and dissolved in 20 mL 0.1 M HCl. The plutonium strip solution was evaporated to about 1–2 mL and taken up with 20 mL 0.1 M HCl. 100 mg Mohr’s salt was added to the solution. The neptunium alpha source was prepared by adding 100 mg Mohr’s salt to the strip solution. The uranium strip solution was evaporated to dryness with 3 × 2 mL 65% HNO3 and re-dissolved in 20 mL 0.5 M HNO3. 100 mg Mohr’s salt was added. The americium alpha source was prepared directly from the strip solution.
Measurement and spectrum evaluation
Alpha sources were measured with Ortec 576A alpha spectrometer (Ortec, USA) attached to Canberra 8701 ADC, 556 AIM and Genie-2000 spectrum acquisition and analysis software (all Canberra, USA). Typically, overnight counting was performed. Alpha spectra were evaluated according to isotope dilution principle. Chemical recoveries were determined from the measured tracer activity compared to the initial activity added to the samples before processing.
Results and discussion
The goal of the work was to develop a simple, robust, rapid and selective separation method of actinides from water samples followed by their alpha spectrometric determination. According to our concept, actinides were pre-concentrated from water samples by precipitation. After dissolution, adjustment of acidity and oxidation state, the actinides were separated by extraction chromatography. Extractants of high distribution coefficients for all actinides and of high selectivity for individual actinides were favored to minimize the actinide leakage during loading and the cross-contamination by other actinides during stripping, respectively. From the commercially available EC resins we considered the selection of TEVA, and the combined TK221 resin containing CMPO and DGA. We use stacked cartridges with TEVA at the top and TK221 resin at the bottom.
Tetravalent actinides (Th(IV), Pu(IV), Np(IV)) have high affinity for TEVA resin. In 3 M nitric acid the capacity factors (k’) for Pu(IV) and Np(IV) are very high, 30,000 and 5000, respectively, while k’ for Th is about 400, also more than enough to retain Th from a load solution of hundreds of mL volume on a 2 mL resin cartridge. On the other side trivalent (Am, Cm) and hexavalent (U(VI)) actinides are poorly retained on TEVA (k’ < 10) what can be regarded as an advantage from the point of selectivity: on TEVA resin only tetravalent actinides are retained. (The capacity factors of actinides on TEVA resin from HNO3 and HCl solutions were determined by Horwitz et al. [2]).
After loading the actinides from 3 M HNO3 on TEVA resin, thorium can be stripped with 9 M HCl while Pu(IV) and Np(IV) are retained. Pu and Np can be stripped together with dilute HCl, as it is often done according to the literature. This part of our method is not different from the "Rapid column extraction method for actinides and 89/90Sr in water samples” developed by Maxwell in 2006 [13]. We wanted to strip Pu and Np sequentially by changing the oxidation state of Pu. We used 9 M HCl/0.03 M TiCl3 to strip Pu(III) while Np(IV) is still retained. Finally, it can be stripped with dilute HCl.
The capacity factors of all actinides on TRU resin are higher than on TEVA resin, from 3 M HNO3 between 30,000 and 300,000 for Th, Np(IV) and Pu(IV), about 4000 for U(VI) and 100 for Am [1]. Therefore Th, Np(IV), Pu(IV) and U(VI) can be strongly retained on TRU resin while the retention of Am is limited (especially in the presence of Fe(III) ions), and the separation of the actinides from each other is challenging.
The situation is similar using DGA resin, where the capacity factors for Th, Np(IV), Pu(IV) and Am from 3 M HNO3 are very high, between 1000 and 40,000, but k’ of U is somewhat less than 100 [9]. Therefore Th, Np(IV), Pu(IV) and Am can be strongly retained on TRU resin while the retention of U(VI) is limited. It was obvious that the combination of TRU and DGA resins in the new TK221 resin could give an appropriate tool for the retention of all actinides including U(VI) and Am.
I. Determination of the distribution ratios
First, we tried to estimate the expected actinide retention on TK221 resin by calculating the k’ values for all actinides by assuming that TK221 resin is a simple mixture of TRU and DGA resins. The individual k’ values on TRU and DGA resins were combined according to the given mixing ratio of the two resins, taking into account the higher extractant loading in TK221. By this approach we assumed that there was no interference between the extractants in the resins. The obtained capacity factors as a function of HNO3 and HCl concentrations are shown as solid lines in Figs. 1 and 2, respectively.

Capacity factors of actinides as a function of nitric concentrations on TK221 resin. Solid lines: k’ values were calculated from the combination of the k’ values measured on TRU [1] and DGA-N resin [9] by Horwitz et al.; Dashed lines: k’ values were experimentally determined by batch uptake experiments on TK221 resin

We also determined the distribution ratios (Dw) of actinides on TK221 from various nitric acid solutions by batch uptake experiments as described in the section Method I. The experimentally determined k’ values were obtained from the measured Dw values and plotted as dashed lines in Fig. 1.
From Fig. 1 it is seen that all actinides can be strongly retained on the TK221 resin from 3 M HNO3 load. The calculated k’ values were between 1000 and 200,000, while the experimental determined values were around 1000 for each tested actinide. The significant difference indicates an interaction between the resin components in TK221, but the nature of the interaction has not been studied. The big advantage of TK221 compared to individual TRU and DGA resins is that all k’ values including those of U and Am became higher than 1000 assuring a strong retention of all actinides from 3 M HNO3 load.
When TK221 resin is attached to TEVA resin, then only the hexavalent U(VI) and the trivalent actinides (Am, Cm) are retained on the TK221 resin. From the estimated k’ values of actinides in HCl solutions (according to Fig. 2) we concluded that the column can be rinsed with 4 M HCl probably without loss of any actinides (if the oxidation states are kept constant) and Am can be stripped with 0.25 M HCl. According to Fig. 2 U(VI) cannot be effectively removed from the resin with dilute acid, therefore 0.1 M ammonium bioxalate as a complexing agent was selected to strip U.
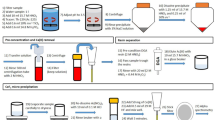
The proposed separation scheme is shown in Fig. 3.

Flowchart of the procedure with Pu-Np separation
Our concept is similar to that of Maxwell et al. [13] but the original three-column method is simplified by using the TK221 resin cartridge instead of stacked TRU and DGA columns. The use of Al(NO3)3 and the redox state adjustment in the load (sulfamic acid/ascorbic acid/NaNO2 to adjust Pu(IV), Np(IV) and U(VI)) are the same. Because of the use of the new resin and the separation of Np and Pu on TEVA resin, the elution conditions had to be optimized.
II. Optimization of the elution conditions
To optimize the elution conditions first elution tests on separated TEVA and TK221 cartridges were performed. The behavior of tetravalent actinides on TEVA resin is well known from the literature. The load solution contained 12 mL 3 M HNO3/1 M Al(NO3)3, sulfamic acid, ascorbic acid, NaNO2, 50 mg Ca and 14 mg Fe(II), as described in Method II. The Ca content in the test refers to a water sample of negligible Ca content, where Ca originates from the added Ca phosphate precipitate. After stripping Th with 9 M HCl, Pu and Np (not present in the test) were stripped together with 0.1 M HCl/0.05 M HF/0.03 M TiCl3. Elution chromatograms of all 4 actinides on TEVA column are shown in Fig. 4.

Elution of Th, Pu, Am and U from TEVA cartridge loaded by a test solutions. (Tests were performed with individual tracers and the chromatograms are shown on a common diagram.) The vertical line between the TEVA titles indicates the volume when the stacked TEVA and TK221 cartridges were separated
From Fig. 4 it is clearly seen that Th and Pu fractions are well separated. They can already be stripped with 10 mL eluent and 15 mL gives a very good separation. During loading or rinsing the column some U loss might appear that gets into the 3 M HNO3 rinse, but it does not contaminate the Th fraction.
Additionally we tried to strip Pu and Np separately by eluting Pu as Pu(III) and Np as Np(IV) fluoride complex sequentially with 15 mL 9 M HCl/0.03 M TiCl3 and 20 mL 0.1 M HCl/0.1 M HF, respectively. Load conditions were the same as in the previous test. The test was performed only with Pu tracer because we did not have a Np tracer of high activity. The chromatogram of Pu on TEVA resin is shown in Fig. 5.

Elution of Pu from TEVA cartridge loaded by a test solution supposing Pu-Np separation
Plutonium is completely eluted with 10 mL 9 M HCl/0.03 M TiCl3. No Pu tailing is observed in the 0.1 M HCl/0.1 M HF solution, the so-called Np fraction. Due to the lack of high activity Np tracer, Np yield was not determined during the optimization test. The yield of Np separation will be determined by the tests of spiked water samples.
Loading and elution of Am from TK221 resin was first tested on a single TK221 cartridge with a series of test solutions containing different amounts of Ca, i.e. 0, 50 and 500 mg Ca. The latter situation refers to a sample of high Ca content where additional Ca is used to form phosphate precipitate to pre-concentrate actinides, like in case of seawater. Americium was eluted with 0.25 M HCl. The chromatograms are shown in Fig. 6.

Elution of Am from TK221 cartridge with 0.25 M HCl as a function of the Ca content of the load (brown color indicates the “U elution”)
The increasing Ca content in the load causes a delay in the elution peak of Am (Fig. 6). In case of a water sample of high Ca content such as 1 L of sea water, 35 mL of 0.25 M HCl is needed to strip Am without tailing into the U fraction. We do not know the mechanism of Am-Ca interference on TK221 resin, but we have observed the anomalous retention of Am in the presence of Ca on DGA resin, as well [18]. We also tested the effect of Fe content on the elution of Am and found no detectable variation on the chromatograms.
Finally, we performed a series of tests with spiked test solutions under the same conditions (Method II.) using stacked TEVA and TK221 cartridges, and measured the actinides eluted from the separated TK221 resin. The chromatograms are shown in Fig. 7.

Elution of actinides from TK221 resin after separation from TEVA resin
According to the chromatogram, Am and U were well separated on TK221, their strip solutions were contaminated neither with Th nor with Pu.
The results of the optimization tests to separate Th, Pu, Am and U are summarized in Table 2. The recoveries of the actinides in their strip solutions were high (≥ 97%), and the cross-contamination was small (< 1%) (the decontamination factors were > 100 for all actinides).
The results of the test experiments suggest that a small amount of uranium (< 1%) may leak into the strip solutions of other actinides, 0.3, 0.2 and 0.8% of U was detected in the Th, Pu and Am fractions, respectively. A few percent U leakage appears during rinsing (and probably loading the columns) reducing slightly the U recovery but is still high (97%). A small leakage of Am (0.7%) in the U strip solution was also observed. However, the small amount of leakage could not be detected in the alpha spectra of spiked water samples (see in the next chapter).
III. Analysis of actinides in water sample
The method was tested by the analysis of tap and sea water samples. 800 mL acidified samples were spiked with a mixture of 230Th, 233U, 239Pu, 241Am and 237Np tracers in order to determine the chemical recoveries in the whole procedure including pre-concentration, separation and alpha source preparation.
Pre-concentration of actinides
was performed by co-precipitation with calcium phosphate as described in Method III. The precipitate is formed from the alkaline earth content of the water and the added Ca carrier (50 mg) with the diammonium hydrogen phosphate after pH adjustment to about 7. Typically, the total Ca amount is between 50 and 500 mg in case of 1 L water (tap or sea) and the amount of phosphate is calculated to be quasi-stochiometric or in small excess so that enough precipitate is formed to carry the actinides. The precipitate after filtration was destructed by evaporation with 65% HNO3 to remove organic contamination.
Load preparation
The evaporation residue was dissolved in 3 M nitric acid/ 1 M aluminum nitrate solution.
The oxidation states of Pu, Np were adjusted to tetravalent which is necessary for their retention on TEVA. First a small amount of iron(III) nitrate (14 mg Fe) is added to the solution to accelerate the reduction of Pu and especially that of Np after the addition of sulfamic acid and ascorbic acid as reducing agents. After the reduction step, Th(IV), U(IV), Np(IV), Pu(III) and Am(III) are present. The solution is cooled in a water bath to room temperature and the redox state is adjusted by the addition of NaNO2. The role of the Na-nitrite is to oxidize Pu(III) to Pu(IV) and U(IV) to U(VI)). The solution was loaded on the stacked TEVA-TK221 cartridges that were washed with 15 mL 3 M HNO3 to remove salt matrix. Then the stack cartridge was disassembled and the columns were eluted separately.
Sequential elution of Th, Pu and Np from TEVA resin
was performed as described in Method III. As Th chloride forms weak complexes with TEVA it can be stripped first with 9 M HCl, while Pu and Np are still retained. If the major interest is to analyze Pu and Np together then they can be stripped with dilute acid containing some reducing agent, such as 0.1 M HCl/0.05 M HF/0.03 M TiCl3. If Pu and Np have to be separated from each other then Pu is stripped after on-column reduction to Pu(III) using Ti(III) with 9 M HCl, while Np(IV) is still retained by TEVA resin. Finally Np can be stripped with 0.1 M HCl/0.1 M HF mixture. Np does not retain on the TEVA resin in dilute acidic media.
Sequential elution of Am and U from TK221 resin
The column was washed with 4 M HCl/0.2 M HF in order to remove Th, Pu and Np potentially leaking from the TEVA resin. Americium was stripped–according to the optimized elution conditions—with increased amount (30 mL) of 0.25 M HCl solution. Uranium was stripped as oxalate complex with ammonium bioxalate solution.
Preparation of alpha sources
Since the acid concentration of the Th and Pu strip solutions (9 M HCl) were considered too high for their co-precipitation with NdF3, the strip solutions were evaporated gently to a volume of about 1 mL, and 20 mL of 0.1 M HCl were added. If the Pu source is prepared directly from 9 M HCl, then Pu losses as high as about 50% occur. Uranium strip solution was evaporated and wet ashed with nitric acid to remove oxalate, then the residue was dissolved in dilute nitric acid. Mohr’s salt was added to the pre-treated Pu, Np and U strip solutions to adjust the oxidation state to (III)/(IV) which is favorable for co-precipitation with NdF3.
Results
The recoveries of the tracers from the water samples are shown in Table 3. Yields are fairly high both from tap (70–100%) and seawater (59–93%) samples, although recoveries from seawater are definitely lower showing some matrix effect. Cross-contamination of other actinides in the alpha sources could not be detected.
I. Method validation
The method discussed above was applied to analyze a water sample within the IAEA-TEL-2021–03 WWOPT proficiency test. The results of 239Pu, 241Am and 244Cm fulfilled all the criteria of acceptance. The results are shown in Table 4.
Conclusions
A rapid method was developed and tested for the separation of actinides from water samples using stacked TEVA and TK221 columns. The Dw values for actinides on TK221 resin determined from HNO3 and HCl media. The method was optimized with model experiments and tested with tap water and seawater samples. High recoveries were obtained for all actinides. Cross-contamination between actinides was not detected. The method was successfully validated by participating in the IAEA-TEL-2021 international proficiency test. Detection limits of the actinides are around 0.1 Bq L−1. Counting time is about 60,000 s. The whole procedure can be completed within 24 h. The procedure is considered suitable for automatization that we are working on.
References
Horwitz EP, Martin KA, Diamond H, Kaplan L (1986) Solvent Extr. Ion Exch 4(3):449–494
Horwitz EP, Chiarizia R, Dietz ML (1992) Solvent Extr. Ion Exch 10:313
Horwitz EP, Dietz ML, Chiarizia R, Diamond H (1992) Anal Chim Acta 266:25–37
Horwitz EP, Dietz ML, Chiarizia R, Diamond H, Maxwell SL, Nelson MR (1995) Anal. Chim. Acta 310:63–78
Kim KG, Burnett WC, Horwitz EP (2000) Anal Chem 72:4882–4887
Thakkar AH (2001) J Radioanal Nucl Chem 248:453–456
LaRosa JJ, Burnett WC, Lee SH, Levy-Polomo I, Povinec PP (2001) J Radioanal Nucl Chem 248:765–770
Ayranov M, Krachenböhl U, Sahli H, Röllin S, Burger M (2005) Radiochim Acta 93:249–257
Horwitz EP, McAlister DR, Bond AH, Barrans RE (2005) Solvent Extr. Ion Exch 23:319–344
Maxwell SL (2006) J Radioanal Nucl Chem 267(3):537–543
Maxwell SL, Culligan BK, Hutchison JB, Utsey RB, McAlister DL (2014) J Radioanal Nucl Chem 300:1175–1189
Maxwell SL, Culligan BK (2009) J Radioanal Nucl Chem 279(3):901–907
Maxwell SL, Culligan BK (2006) J Radioanal Nucl Chem 270(3):699–704
Maxwell SL (2011) Anal Chim Acta 701(1):112–118
Maxwell SL, Culligan BK, Hutchinson JB (2014) J Radioanal Nucl Chem 299(3):1891–1901
Maxwell SL, Culligan BK, Noyes GW (2010) J Radioanal Nucl Chem 286(1):273–282
Eichrom’s Analytical Procedure ACW16VBS (2019) https://www.eichrom.com/wp-content/uploads/2018/02/ACW16-12_Am-Pu-U-Np-Th-Water-VBS.pdf Accessed 16 December 2021
Eichrom’s Analytical Procedure ACW17VBS (2019) https://www.eichrom.com/wp-content/uploads/2018/02/ACW17-13_Am-Pu-U-Np-Th-Sr-Water-VBS.pdf Accessed 16 December 2021
LaRosa JJ, Burnett W, Lee SH, Levy I, Gastaud J, Povinec PP (2001) J Radioanal Nucl Chem 248:765–770
LaRosa JJ, Gastaud J, Lagan L, Lee SH, Levy-Polomo I, Povinec PP (2005) J Radioanal Nucl Chem 263:427–436
Maxwell SL (2008) J Radioanal Nucl Chem 275(3):497–502
Thakur P, Ballard S, Conca JL (2011) J Radioanal Nucl Chem 287:311–321
Harrison JJ, Zawadzki A, Chisari R, Wong HKY (2011) J Environm Radioact 102:896–900
Qiaou J, Hou X, Szeier P, Golser R (2013) Anal Chem 85:11026–11033
Dai X, Kramer-Tremblay S (2014) Anal Chem 86:5441–5447
Habibi A (2015) Anal Chim Acta 883:109–116
Wang Zh (2018) J Radioanal Nucl Chem 315(1):103–110
Vajda N, Torvenyi A, Kis-Benedek G, Kim CK, Bene B, Macsik Z (2009) Radiochim Acta 97(8):395–401
Vajda N, Zagyvai M, Groska J, Molnar Zs, Bokori E, Braun M (2020) J. Radioanal. Nucl. Chem. 326:695–710
Acknowledgements
The work was performed with the professional support of the doctoral student scholarship program of the co-operative doctoral program of the ministry of innovation and technology financed from the national research, development and innovation fund. Contract ID: RH/527-14/2021.
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Papp, I., Vajda, N. & Happel, S. An improved rapid method for the determination of actinides in water. J Radioanal Nucl Chem 331, 3835–3846 (2022). https://doi.org/10.1007/s10967-022-08389-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08389-9