
Abstract
Development of 3H, 14C, 41Ca, 55Fe and 63Ni radiochemical analysis methods were carried out independently by two laboratories using both inactivate and activated concrete samples. Two preliminary radioanalytical procedures for the non-volatile radionuclides (41Ca, 55Fe, 63Ni) and one Thermal oxidation method for the volatile radionuclides (3H, 14C) were developed. The difficulties in the method development and analysis of results are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiochemical analysis of Difficult To Measure radionuclides (herein referenced as DTM) in decommissioning waste has been a challenge for decades. DTMs are a general term for alpha and beta emitters, which require their quantitative extraction from the matrix and purification from other radionuclides. Destructive analysis techniques are needed for the determination of DTMs such as 3H, 14C, 41Ca, 55Fe and 63Ni in solid decommissioning waste. In general, the radionuclides of interest are first separated from the matrix, then separated and purified from other radionuclides and, finally, measured using liquid scintillation counting (LSC). Other measurement techniques (e.g., X-ray, proportional counters, and mass spectrometry) are available but not as often utilised [1]. Due to the long separation and purification processes, the yields of the analytes of interest are corrected in the end of the procedure by using analytical measurement techniques. Also, standard addition method, in which radioactive standard is added and analysed, can be used. 3H and 14C yields are determined experimentally using standard solutions, because utilisation of corresponding stable elements (e.g., stable 56Fe for 55Fe) is not viable due to the difficulties of 1H and 12C analysis.
One of the most important material in decommissioning projects is concrete, because contaminated and activated concretes are a major source of radioactive waste volume. These two types of concretes differ in their nuclide vector due to the source of radionuclides. The contaminated concretes can contain variety of radionuclides depending on the origin. For example, the contaminated concrete can originate from a waste silo, into which laboratories working on variety of radionuclides have collected their contaminated wastewaters or from a floor of a dedicated radiochemistry laboratory, in which only specific radionuclides have been handled. In the first case, continuous contamination can be expected whereas in the second case, the contamination can be unevenly distributed. In activated concrete, the radionuclide vector depends on the original chemical composition of the concrete, irradiation history and distance from the irradiation source. Additionally, the activated concrete can be divided to heavy and normal concrete. Heavy concrete mainly consists of BaSO4 and silicates giving the material high absorption capacity for gamma radiation i.e., shielding properties are increased compared to normal concrete [2]. In this case, the heavy concrete contains higher amounts of Ba, which activates to gamma emitting 133Ba whereas in normal concrete, there is relatively more Ca, which activates to 41Ca decaying with electron capture. Therefore, complete separation of 133Ba from the 41Ca fraction is especially important when activated heavy concrete is analysed. Additionally, acid digestion even with HF cannot completely decompose heavy concrete [2]. The concrete can be also a mixture of contaminated and activated concrete and as such, the different types of concretes can require different approaches in the sample collection and analysis. For example, tritiated water (HTO) contaminated structures require special consideration regarding the volatility of 3H during sampling whereas high temperatures are needed in 3H analysis of activated concrete, in which 3H is partly strongly bound in the lattice structures [3, 4].
One challenge in determining DTMs from concrete is sample digestion, which depends on the particular concrete composition and scope of the study, i.e., whether acid digestion gives adequately dissolved sample releasing the isotopes of interest into solution, or if total dissolution of the sample, including silicates, is required. Both acid digestion and alkaline fusion have benefits and drawbacks and therefore, it is not always clear, which one is the better option for the particular purpose [5]. Sometimes the choice of dissolution method is based on familiarity, safety, easiness, availability and time resources. Additionally, almost quantitative dissolution can be enough, even though some more efficient method would exist. A variety of alkaline fusion methods exist for a complete dissolution of, e.g., geological, environmental and nuclear decommissioning samples. These methods utilise sodium hydroxide [6, 7], lithium borate [8, 9], sodium peroxide [10, 11] and similar alkaline compounds in different mixtures together with high temperature (most often over 600 °C) for complete destruction of even resistant silica minerals. In traditional methods, alkaline fusion is carried out in crucibles which are manually placed in and out of the oven. Manual operation of the extremely hot and alkaline sample vessels causes a health and safety concern for the workers. However, sophisticated and safe instruments have been developed for this purpose during the last decades, which minimise the safety hazards of sample treatment. Modern fusion techniques also enable automated fusion processes [9]. In this study, preliminary tests were carried out according to Ref. [2] using 3 g of NaOH and 1 g of Na2CO3 mixed with up to 5 g of concrete and fusing them in a crucible at 500–550 °C for 3–4 h. Dissolving of the fused cake was attempted with hot solution of 0.2 M Na2CO3 [2]. However, the tests were not successful resulting in dissolving of the porcelain crucible and therefore, different acid digestion methods were tested and their efficiency in dissolving Fe, Ni and Ca from the concrete is reported.
Second challenge in determination of DTMs is related to the above mentioned alkali fusion tests in which repeating of published method was not successful. In this case, fairly simple method was repeated several times, but the results were not satisfactory. The difficulties in repeating of published methods is also discussed in the experimental section of this paper as the two laboratories produced different results with utilisation on aqua regia in dissolving of the concrete.
Third challenge is the presence of interfering elements (i.e., radioactive and stable) depending on the matrix, contaminants and activated radionuclides. In the case of matrix, for example large amounts of stable Fe has been shown to cause difficulties in the analysis of activated steel samples due to the need of large amount of ion exchange resin in Fe and Ni separation [12]. The challenge has been addressed by carrying out the analysis with aliquots and preparatory precipitations and by increasing the necessary amount of ion exchange resin [12]. In the case of contaminants and activated radionuclides, the challenge is caused by the similar behaviour of the interfering radionuclides with the analyte of interest. In the case of steel, one of the main interfering radionuclides is 60Co in 63Ni analysis as Co easily follows Ni [13]. This challenge was addressed by utilisation of method by Hazan and Korkisch [13, 14] or with two Ni-resin separations [13]. In this study, the studied concrete was known to contain high amounts of Fe whereas its activity concentrations were known to be low according to activation calculations.
The third challenge can be encountered in the measurement phase. Especially low energy DTMs, such as 55Fe and 41Ca, which decay with electron capture, suffer from quenching in the LSC measurement. Quenching can be lowered with selection of the chemical composition of the purified fractions, e.g., 1 M H3PO4 for 55Fe [13]. Additionally, low activity concentration requiring large amount of sample can cause quenching due to large amounts of stable elements in the 41Ca fraction as shown in this study with analysis of 10 g of activated concrete.
In a nutshell, this paper presents the development of three preliminary radiochemical analysis methods for the determination of 3H, 14C, 41Ca, 55Fe, and 63Ni in activated concrete. The activated concrete originated from the biological shield concrete of FiR1 TRIGA Mark II type research reactor in Finland. The radiochemical analysis methods were first tested with inactive FiR1 biological shield concrete and then modified for activated FiR1 biological shield concrete analysis. Acid digestion of the concrete followed by precipitations, ion exchange and extraction chromatography column separations were utilised for the measurement of 41Ca, 55Fe and 63Ni. The volatile 3H and 14C were analysed using the thermal oxidation method. The encountered difficulties in the DTM analyses (e.g., solubility, interfering radionuclides, quenching) are discussed in the experimental section and the problem solving were carried out independently by the two laboratories. The consequences of the made decisions are also discussed.
FiR 1 biological shield concrete
The on-going characterisation efforts of FiR1 TRIGA Mark II type research reactor have been published previously [15,16,17]. The characterisation of the biological shield concrete began with drilling of core samples from the non-activated part of the biological shield concrete in order to determine the chemical composition of major activating elements for activation calculations. The summary of relevant elements, which produced DTMs analysed in this study in FiR1 biological shield concrete, are presented in Table 1. The analysis results are an average of three replicate measurements with standard deviation. The second sampling campaign was carried out in a few years later order to collect activated core samples.
Experimental
Two radiochemical method developments (Method 1 and 2) for the analysis of 41Ca, 55Fe and 63Ni were carried out independently by two laboratories, using inactive concrete samples and analysis of the purified fractions using elemental analysis. Method 3 was developed using inactive concrete samples spiked with liquid 3H and 14C standards. All the methods were different combinations of published articles [2,3,4, 12,13,14, 19,20,21,22,23,24,25,26,27,28,29]. The Methods 1 and 2 were both based on acid digestion, precipitations, ion exchange and extraction chromatography whereas the Method 3 was based on thermal oxidation using a combustion furnace. In all cases, the initial tested methods with inactive concrete required further developments for the radiochemical analysis of activated concrete. The procedures and actions taken are discussed in the subsections.
Sample preparation
A dedicated drilling configuration was developed for the sampling of the inactive and activated concrete cores. In brief, the sample was drilled with a hollow drill and the powdered concrete was collected. The powdered concrete had a small particle size, which was foreseen to enable more efficient complete destruction of the material in acid digestion due to larger surface area and also better release of volatile DTMs in the thermal oxidation technique as suggested in Ref [12].
Chemicals and equipment used in Method 1
All chemicals and reagents (Na2CO3, NaOH, NH4Ctr i.e., ammonium citrate, Ni(NO3)2·6H2O, CaCl2·4H2O, FeCl3·6H2O, CoCl2) were of analytical grade or purer. Solutions were prepared into deionised water. Bases and acids were of analytical grade with concentration of 85 w-% H3PO4, 25 w-% NH4OH, 65 w-% HNO3, 36–38 w-% HCl, 72 w-% HClO4. Ion exchange columns were prepared using AG 1 × 4 50–100 mesh anion exchange resin (BioRad) and extraction chromatography columns using Ni-Resin B 100–150 µm (Triskem International).
Orion 2 Star pH Benchtop pH meter combined with Ross combination pH electrode (Thermo Scientific) was calibrated with pH 4, 7 and 10 AVS TITRINORM buffer solutions.
41Ca, 55Fe and 63Ni were analysed using HIDEX 300 SL liquid scintillation counter (later referred as Hidex) with TDCR (triple to double coincidence ratio) technology for the counting efficiency determinations. Samples were mixed with OptiPhase HiSafe 3 (later referred as HiSafe) liquid scintillation cocktail purchased from Perkin Elmer and let to stabilise at least 12 h before measurement in order to minimise luminescence. 1 ml or more of 41Ca, 55Fe and 63Ni sample solutions were mixed with 10–20 ml of HiSafe.
Elemental analysis of the acid digested solution and radiochemical yield analyses were carried out using Agilent SVDV 5100 ICP-OES (Inductively Coupled Plasma Optical Emission Spectrometry). The samples were diluted into 1% suprapur HNO3 solution. Ca, Fe and Ni measurements were carried out using radial view with four different wavelengths in order to detect interference. Since all four wavelengths gave consistent results, the reported results were taken from the first selected line, namely 315.887 nm for Ca, 234.350 nm for Fe, and 216.555 nm for Ni. Ca, Fe, and Ni standard solutions were prepared using IV-Stock-4 (Inorganic Venture, USA) 1000 ppm multielement standard.
Chemicals and equipment used in Method 2
All used reagents were of analytical grade and reagent solutions were prepared in Milli-Q-water. Acid and base stock solutions were 85 w-% H3PO4, 65 w-% HNO3, 36–38 w-% HCl, 25 w-% NH4OH, and 48 w-% HF. Ion exchange chromatography resin Dowex 1 × 4, 50–100 mesh (Sigma-Aldrich) and extraction chromatography resins TRU, 100–150 µm (Triskem International) and Ni resin, 100–150 µm (Triskem International) were used in radiochemical column separations.
The activity concentration of 41Ca, 55Fe, and 63Ni in the final separated fractions was determined with Quantulus 1220 Ultra Low Level Liquid Scintillation Counter (Perkin-Elmer, previously Wallac). 18–19 ml of LSC cocktail Ultima Gold uLLT (Perkin-Elmer) was added to each sample (1–2 ml of sample solution), always having 20 ml of the total LSC sample volume. The sample mixture was shaken and kept 24 h inside the LSC counter before starting the measurement, to avoid interference from chemiluminescence. Counting efficiency of Quantulus 1220 for 55Fe (Eβ = 231 keV, Ex-ray = 5.89 keV), 63Ni (Eβ = 66.95 keV) and 41Ca (Ex-ray = 3.31 keV) was determined by measuring quench series samples, prepared from 3H (Eβ = 18.6 keV), 63Ni and 41Ca standard solutions, respectively. The obtained efficiency/SQP plots were used for quench correction in activity calculation of the real samples.
Chemical yield of Ca, Fe, and Ni in the separated Fe and Ni fractions was determined with Agilent 4100 MP-AES (microwave plasma-atomic emission spectrometer). The selected emission wavelengths were 393.366 nm for Ca, 371.993 nm for Fe, and 345.846 nm for Ni. Calibration standard series for MP-AES were prepared from 1000 ppm Fe and Ni reference solutions (Romil) and from ERM-AE701 Ca standard solution (JRC, Geel, Belgium). All measurement samples were prepared by diluting the samples with 5% suprapur HNO3 (v/v).
Chemicals and equipment used in Method 3
0.1 M HNO3 trapping solution for 3H was prepared into deionised water and 65 w-% HNO3 (analytical grade). Carbo-Sorb® E (later referred as Carbosorb) CO2 absorber for 14C was purchased from Perkin Elmer. Expired 3H and 14C standards were dissolved into deionised water obtaining 70 000 dpm/ml and 42 000 dpm/ml solutions, respectively. These standards were considered to be fit for purpose as the calculated yield results were produced with comparison to original solutions.
The combustion furnace was a Pyrolyser-2 Trio (RADDEC International), which enabled simultaneous analysis of two samples. The Pyrolyser had three furnaces and the samples were placed in the first furnace. The temperature in first furnace was controlled by the operator. The second furnace had been set by the manufacturer to heat to 500 °C as soon as the first furnace reached 500 °C. The second furnace ensured that volatile organic species were efficiently transferred into the catalyst zone, i.e., third furnace. The third furnace was pre-heated to 800 °C as set by the manufacturer. The catalyst (0.5% Pt-alumina) was placed in the third furnace in order to maximise the 3H conversion to tritiated water (HTO) and 14C to CO2. The HTO and CO2 were trapped into 20 ml trapping solutions. In this study, 0.1 M HNO3 and Carbosorb trapping solutions were used even though also 0.4 M NaOH for 14C trapping was tested. Some evaporation occurred and therefore it was controlled with weighing the solutions before and after the heating cycle. The airflow (compressed air) was adjusted to 0.2–0.25 l min−1 and O2 (industrial grade) 50:50 flow was added when the first furnace reached 500 °C while keeping the same flow rate as previously.
The same LSC and scintillation cocktail were used as in Method 1. 5 ml of 3H and 14C sample solutions were mixed with the liquid scintillation cocktail and let to stabilise at least 12 h. The recommended liquid scintillation cocktail for Carbosorb is Permafluor E + by Perkin Elmer. However, HiSafe gave consistent results even though it occasionally formed a gel with Carbosorb.
Testing of Method 1 in inactive concrete and the radiochemical analysis of 41Ca, 55Fe and 63Ni in activated concrete
The initial radiochemical Method 1 for the determination of 41Ca, 55Fe and 63Ni was a combination of published articles [2, 12, 25, 26]. The procedure was based on 4 h acid leaching of the solid subsample (0.5 g) using 30 ml of conc. HCl and 10 ml of conc. HNO3, removal of the undissolved solid via filtering through a glass filter, and separation of Ca from metals (e.g., Fe and Ni) and transuranics using hydroxide precipitation [2]. Hold back carriers (4 mg Co) and carriers (Ca to 200 mg, Fe to 4 mg, Ni to 2 mg depending on the original content in the acid digested solution i.e., approximately 175 mg Ca, 2 mg Ni and no Fe) were added after acid digestion. Ca in the supernatant was further purified from other interfering radionuclides (e.g., Ba, Cs, Sr and Ra) using sequential precipitations and dissolving of the precipitate [2]. Fe and Ni were separated from each other and purified similarly to the Radioanalytical Method 1 in Ref [12] using AG ion exchange and Ni-resin.
The Method 1 was tested with three 0.5 g subsamples of the inactive concrete. The efficiency of the acid leaching to release the Ca, Fe and Ni from the solid matrix was determined by analysing the Ca, Fe and Ni concentrations in the acid leached solutions and comparing the results with the previously determined concentrations in Table 1. The results (M1a-c) in Table 2 show that the acid leaching was able to release 50–60% of the original Ca content and 60–80% of the original Fe content. The Ni concentrations were below limit of the detection. Even though the solubility of the concrete was foreseen to be a major challenge, several references indicated that in the case of normal concrete (e.g., not heavy concrete), complete destruction of the matrix was not necessary since leaching in aqua regia was able to release more than 98% of the Ca [2]. As the measured results were contrary to this statement, Method 1 was modified in order to achieve complete destruction of the matrix using HNO3, HF and HClO4 acid mixture [25]. The change of acid digestion mixture from HNO3 and HCl mixture to 5 ml conc. HNO3, 5 ml conc. HF and 3 ml conc. HClO4 and subsequent treatment with 5 ml conc. HNO3 and 3 ml conc. HClO4 mixture was successful as the yields in the acid digestion of 0.6 g activated concrete subsamples M1d-f was increased from 50 to 60% and 60–80% to 80–82% and 82–86% for Ca and Fe, respectively. The destruction of the matrix was considered to be complete even though small amount of solid silicates remained as the acid mixture was evaporated.
The yields in the purified Ca, Fe, and Ni fractions in the inactive (M1a-c) and activated (M1d-f) concrete subsamples are presented in Table 3. As the Ca yield results show, the sequential precipitations and dissolving of the Ca fraction ended up in low yields of less than 18% in the inactive concrete samples. Due to the easiness of sequential precipitations, almost the same method was continued also in the analysis of the activated concrete subsamples, the difference being longer reaction times in the precipitation steps. Even though the results in Table 2 show that Ca yields in the purified fractions of activated concrete were increased compared to the corresponding results using in inactive concrete, the yields were still relatively low i.e., 24–34%.
The Fe yield results in the purified Fe fractions of inactive concrete were also relatively low (48–80%) most likely due to the high Fe content i.e., AG resin capacity for Fe had been breached. Therefore, the amount of AG resin was increased from two 10 g AG resin columns to one 30 g AG resin column in the analysis of the activated concrete. However, the change of two subsequent 10 g AG resins to one 30 g AG resin was not successful to increase the Fe yield in the purified fractions as the yields lowered from 48 to 80% to 13–19%.
As discussed in previous publications [12, 14, 22, 27], purification of Ni from Co can be challenging. The yield results for Ni in inactive concrete subsamples show that about 90% of the Ni was carried over to the final purified Ni fraction. The above mentioned modifications (i.e., acids in acid digestion, longer reaction times in Ca precipitations, and amount of AG resin in Fe and Ni separation) were not targeted for Ni. However, the Ni yield was increased to near 100% in the activated concrete samples with the modifications. The final Method 1 utilised in the radiochemical analysis of the activated concrete samples is presented in Fig. 1.

Radioanalytical Method 1 for determination of 14C, 55Fe and 63Ni in activated concrete
In all the cases, the 41Ca, 55Fe, and 63Ni activity concentrations in the active concrete subsamples M1d-f were below limits of detection (Table 4). The limits of detections were calculated using the Currie method [30]. In the Finnish regulations by the Radiation and Nuclear Safety Authority [31], the free release limits for 55Fe and 63Ni are 1000 Bq g−1 and 10 Bq g−1, respectively whereas there is no release limit given for 41Ca. On the other hand, the free release limits for 45Ca and 47Ca are 100 Bq g−1 and 10 Bq g−1, respectively. Therefore, it can be concluded that the provided results are well below the free release limits whereas scientifically speaking, the method could be tested further in order to increase the yield or larger sample size could be used in order to overcome the low yields. Larger sample size, however, can introduce further problems such as significantly higher amount of stable Fe in the samples complicating the AG resin separation as discussed in next sub-section.
LSC counting efficiencies for 41Ca, 55Fe, and 63Ni by TDCR with CoreF correction (Hidex) are presented in Table 4. CoreF correction is a mathematical corrective function for deviations of TDCR at higher quench levels. The CoreF corrections for 55Fe and 63Ni were provided by the manufacturer whereas lack of corresponding correction for 41Ca resulted in use of CoreF correction of 55Fe which similarly decays with electron capture. The obtained values had some variation among subsamples, from 0.317 to 0.649, and there was no clear difference between efficiency values for different radionuclides.
Testing of Method 2 in inactive concrete and the radiochemical analysis of 41Ca, 55Fe and63Ni in activated concrete
Method 2 was modified from previously published methods for separating Ca, Fe and Ni from each other and from disturbing elements by hydroxide and carbonate precipitations and extraction chromatography [2, 22,23,24, 28]. Three parallel subsamples of 0.5 g inactive concrete were used for experimenting the initial Method 2. 20 mL of aqua regia was added to each concrete sample and the samples were digested on a hot plate for four hours. The sample solutions were filtered and solution parts were evaporated to dryness. After treatment with 2 ml of conc. HCl, the evaporation residues were dissolved to 1 M HCl and Ni carrier (2 mg) was added to each sample. Small fractions were taken from the sample solution before and after addition of Ni carrier for MP-AES measurements. The results in Table 2 show that the sample dissolution procedure with aqua regia released 85–94% of Ca and > 97% of Fe from the inactive concrete subsamples to the solution. The percentages were calculated similarly with Method 1, i.e., from the ratio of the determined concentrations in digestion solution to the previously determined reference values in Table 1. Concentration of Ni was below the detection limit in the subsamples of 0.5 g. Visually estimating, approximately half of the concrete was dissolved to acids during leaching. Although the dissolved amounts of Ca and Fe were satisfactorily high and the residual concrete contained probably mainly silicates, for the next tests with much higher sample masses of active concrete, a more efficient digestion method was sought. Therefore, sample dissolution method was changed from a single aqua regia digestion to a more extensive procedure. As discussed with Method 1, the activated concrete contained only low amounts of investigated radionuclides and for obtaining results exceeding detection limits, a large subsample mass was needed for the radiochemical analysis. The main reason for adjustments was much higher subsample amount (10 g) required for activated concrete, compared to small subsamples (0.5 g) used with inactive concrete. The concrete samples were first treated with 9 M HCl heating on a hot plate for 30 min, evaporated to dryness and a series of HF attacks (milliliters of HF was added to samples, heated and evaporated to dryness) was followed. The use of HF was expected to improve dissolution of residual silicates in the concrete samples. Finally, the samples were digested with a mixture of HNO3:HCl:HF (10:8:2) for three hours on a hot plate and evaporated to dryness. Despite extensive leaching procedure, only ~ 60% of concrete was dissolved, the remaining 40% being chemically resistant silicates. The dissolved fraction might have been increased with even more HF treatment steps.
In the inactive concrete subsamples, Ca was separated from Fe and Ni in hydroxide precipitation and Ca fraction was further purified with carbonate co-precipitation and two different anion exchanges. Fe and Ni were separated from each other with TRU column separation and Ni fraction was further purified with Ni resin column. For later tests and analyses of the activated concrete, efficient purification cycles were planned in order to remove interfering radionuclides from the final purified fractions. In a previous study [24], Ca fractions were treated with (at least) three carbonate precipitations and 2–3 anion exchange column pairs in order to efficiently purify 41Ca fraction. Additionally, based on previous experiences with activated steel containing higher concentration of 60Co compared to concrete [13], Ni fractions of activated concrete would require at least two Ni resin column separations for decreasing the concentration of 60Co to a tolerable level, concerning LSC measurement. Furthermore, the residual activity of 60Co in LSC spectrum of 63Ni (and 60Co) could be taken into account by measuring exact activity of 60Co from the LSC sample by gamma spectrometry [13].
The yields of Ca, Fe and Ni after chemical purification of inactive concrete samples M2a-c were 43–61% for Ca, 29–36% for Fe and 92–104% for Ni (Table 3). For Ni, the separation method can be considered successful, whereas for Fe and Ca did not work as well. Explanation for low Fe yield is the loss of Fe in TRU separation: part of Fe in the sample load solution did not bind to TRU resin and it was eluted within the Ni fraction. Fe content of the subsamples exceeded 3 mg of Fe which is considered being the upper tolerance limit of a regular (0.7 g of resin) TRU column for Fe [27]. Therefore, working with even larger sample masses, two options existed for maintaining efficient separation of Fe, Ni and Co from each other, and not exceeding the binding capacity of the resin i.e., (1) it would be possible to use significantly higher amount of TRU resin, or alternatively (2) use ion exchange instead of extraction chromatography for separating Fe, Ni and Co from each other.
The radiochemical separation procedure for analysis of the activated concrete was a combination of methods published by Eichrom [28] and Hou [32] for Fe and Ni and by Ervanne et al. [24] for Ca. For Ca, the analysis method was almost the same as with inactive concrete, but the carbonate precipitation was replaced with an oxalate precipitation, because for some reason carbonate precipitate did not form in the solutions of the activated concrete. Only one anion exchange column cycle (first column purification in 8 M HCl and the second in 8 M HNO3) was performed for Ca fractions, because based on gamma measurements, there was no 60Co or other gamma emitters left in the Ca solutions after the first anion exchange cycle. For Fe and Ni analysis in activated concrete, it was decided to replace TRU column separation with Dowex 1 × 4 anion exchange column separation. Although it would have been possible to perform Fe/Ni/Co separation with a higher amount of TRU resin, it was more economical and less sensitive to column load capacity to use anion exchange resin instead.
The yields of purified fractions in the activated concrete subsamples were 48 and 90% for Ca, 57% for Fe, 72 and 82% for Ni (Table 3). Compared with corresponding yields in inactive concrete, the yields for Ca and Fe were higher, and for Ni the yield was lower in activated concrete. The increase in the yield of Fe was due to replacement of TRU separation with inadequate resin amount with anion exchange separation. Based on testing of Method 2 with inactive concrete and its modification, few hours leaching with aqua regia produced adequate dissolution of concrete, concerning determination of 41Ca, 55Fe, and 63Ni. The final Method 2 utilised in the radiochemical analysis of the activated concrete samples is presented in Fig. 2.

Radioanalytical Method 2 for determination of 14C, 55Fe and 63Ni in the activated concrete
The activity concentrations of 41Ca and 63Ni in the activated concrete were below limit of the detection and the activity concentrations of 55Fe were 2.3 ± 3.7 and 2.9 ± 4.6 Bq g−1 (two sigma uncertainty) in the two subsamples (Table 4). Uncertainties of the activity concentration values for 55Fe were high, due to low activity concentration in the concrete (high statistical counting error of radioactivity) and quenching in LSC measurement, leading to decreased counting efficiency.
The LSC counting efficiencies for 41Ca, 55Fe, and 63Ni by E/SQP plotting (Quantulus) are presented in Table 4. The relationship between beta particle energy and detection efficiency of Quantulus is clearly seen as decreasing efficiency values with decreasing decay energy, from 63Ni via 55Fe to 41Ca. The counting efficiency for 55Fe in two concrete samples containing 15 and 22 mg of stable Fe was 7 and 8%, respectively, whereas it was 32% in an unquenched standard sample. Even though the subsample mass was maximized in order to obtain radioactivity concentrations over detection limit, high concrete amount led to matrix-related quenching with all determined beta emitters, due to precipitation or colour in the samples. This was crucial in detecting 55Fe and especially 41Ca, which decay by electron capture, having very weak Auger or x-ray emissions, 5.9 keV and 0.3–3.6 keV, respectively. The difference in counting efficiencies for these low energy betas obtained by Hidex (three photomultiplier tubes) and Quantulus (two photomultiplier tubes) is remarkable: Hidex had roughly tenfold efficiency for 55Fe and 41Ca, compared to Quantulus. For 63Ni having higher decay energy, the counting efficiencies of the two LSCs were at the same level. This observation indicates Hidex as an attractive option in determination of low energy beta emitters. On the other hand, for samples having very low activity content, Quantulus may be the only choice due to its generally lower background count rate.
Testing of Method 3 in inactive concrete and radiochemical analysis of 3H and 14C in activated concrete
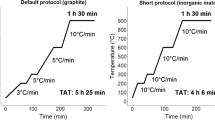
The Method 3 tests using thermal oxidation were carried out using 100 mg of inactive concrete spiked with 100 µl of 3H and 14C standard solutions. Since some of the 3H in activated concrete originates from Li, which can be tightly bound in the lattice, the temperature in the first furnace needed to reach high temperature of 900 °C [3, 4]. Therefore, a heating profile recommended in the manufacturer’s additional training material was adjusted and the heating profile of the first furnace was set to increase 10 °C min−1 until 900 °C and stay at 900 °C for 90 min. 20 ml of 0.1 M HNO3 trapping solution was used for the HTO trapping. Initial tests were carried out with 20 ml of 0.4 M NaOH and 20 ml of Carbosorb solutions for trapping of 14C. The results showed that only 50% of the released 14C was trapped by the 0.4 M NaOH whereas 100% was trapped with Carbosorb. Therefore, Carbosorb was used as the 14C trapping solution. The trapping solutions were cooled with an ice block in order to lower evaporation. Evaporation was determined by measuring the initial and final weights. 5 ml of each trapping solutions were mixed with 10 ml of HiSafe and measured using LSC after stabilisation inside of Hidex at least for 12 h.
The Method 3 was able to provide consistent results for several spiked inactive concrete samples giving an approximate yield of 92% for 3H and 115% for 14C after evaporation correction. The evaporation rates for 0.1 M HNO3 and Carbosorb trapping solutions were approximately 0.2 ml h−1 and 0.6 ml h−1, respectively. Analysis time in the pyrolyser was approximately 3.5 h. The results suggested that some 3H was carried either in or through the Carbosorb absorption solution. Additionally, the over 100% yield of 14C suggested that either 3H was absorbed into the Carbosorb or evaporation of Carbosorb did not cause decrease of 14C. The 14C spectra did not support the theory of 3H absorption and additionally Carbosorb is specific for carbon. Therefore, an additional study was carried out in order to see if evaporation of Carbosorb also released 14C. Four samples with 25 µl of 14C standard solution were mixed with 5 ml of Carbosorb. First sample was closed with a cap in order to minimise evaporation whereas nitrogen gas flow was subjected into the 3 remaining samples in order to evaporate increasing amount of Carbosorb from the vials. In the end of the experiment approximately 8, 17 and 33% of solutions were evaporated. Afterwards 10 ml of HiSafe was added into all the samples and they were measured using LSC after at least 12 h stabilisation. The results showed that 100% of 14C remained in the evaporation vials i.e., even though the Carbosorb evaporates, 14C remains in the solution. Therefore, it was concluded that experimental yield for 3H was 92% (with evaporation correction) and for 14C 100% (without evaporation correction). The 14C activity concentration in the FiR1 activated concrete was below LOD whereas 3H activity concentration was 43 ± 13 Bq g−1 calculated from 10 replicate measurements.
Since spiked samples have different 3H and 14C speciations compared to activated concrete, Method 3 was further studied with increasing amount of FiR1 activated concrete in order to establish linearity range. A linear relation with increasing amount of sample was expected until a plateau. The plateau was expected to represent a situation in which the volume of the sample blocks efficient release of the analytes or a situation in which the absorption solutions are saturated. The studied sample sizes varied from 100 mg to 5 g and the results showed linear correlation between yield corrected DPM and sample mass with R2 0.9935. Linear range may be even higher but larger sample sizes were not studied due to the limited amount of sample.
In general, the radiochemical analysis of 3H and 14C using a pyrolyser was a straight forward process, but several parameters need to work optimally in order to produce reliable results. Firstly, the amount of sample needs to be tested. In case when consistent results are obtained using different amounts of sample, the results are more reliable. Secondly, the heating profile needs to be tested for the sample type. Especially high organic matter content in the sample requires slow increase of temperature in order to avoid uncontrolled combustion of the organic matter i.e., soot. Additionally, in cases when high temperatures are used, cooling time can be long limiting the analysis frequency to be only two samples per day. Thirdly, the flow rate should be constantly in the optimal flow rate range. Especially addition of O2 as the first furnace reaches 500 °C requires good flow in order to oxidise 3H and 14C efficiently. Additionally, too low flow rate can lower the HTO and CO2 to reach the trapping solutions and too high can push them through the solutions too quickly lowering the trapping percentage. Fourthly, the oxidative power of the catalyst is limited and change of the catalyst is recommended after every 15th analysis. However, the oxidative power is affected by the analysed samples and can be decreased towards last analysis. All these above mentioned parameters can occur separately or in different combinations making the radiochemical analysis of 3H and 14C challenging. The effects of these parameters would be detected if tracers could be used for the yield determination. However, use of tracers for 3H and 14C determinations is not possible and determination of an estimated yield needs to be carried out separately. Most often, such as in this study, spiked samples were used even though it was clear that the speciations of 3H and 14C in standard solutions are different to the speciations in activated concrete. Contaminated concrete would be more similar to spiked concrete.
Conclusions
Three radioanalytical methods were tested for determining volatile (3H and 14C) and non-volatile (41Ca, 55Fe and 63Ni) beta emitters from activated concrete samples. Based on the first results with inactive concrete, the methods were further modified for improving their forthcoming performance with activated concrete samples. Comparison of Method 1 inactive and activated subsamples’ yields for both Fe and Ca increased when complete destruction of the matrix was obtained. On the other hand, in comparison of Method 2, inactive and activated subsamples show significant decrease in Fe and Ca yields, which are most likely caused by the significantly higher amount of sample under digestion. Formation of CaF2 from Ca in concrete and F in added HF is also possible in some extent, this would have decreased effect on Ca yield. Comparison of Method 1 and 2 in inactive concrete subsamples shows significantly different results even though both methods used same amount of sample, but different volumes of aqua regia and digestion time. On the other hand, digestion temperatures were not recorded. As the utilised Methods 1 and 2 did not significantly differ, the reason for better yields using Method 2 remained unknown emphasizing that even published methods can produce different results depending on the laboratory.
The radiochemical analyses of activated concrete suffered from low activities and color quenching in LSC. Both 41Ca and 63Ni activity concentrations were below limit of detection even in cases in which 10 g subsamples were analysed. 55Fe activity concentrations were below limit of detection with 0.6 g subsamples and suffered from high uncertainties with 10 g subsamples. Out of the volatile DTMs, 14C activity concentration was below limit of detection whereas consistent results were obtained for 3H.
Later the same activated concrete material was analysed among wider group of laboratories and the obtained experiences and results are summarised in Refs. [33, 34]. Comparison of the results presented in this paper are well comparable with the results of the wider group of laboratories as no major difficulties were observed for 3H analysis, thermal oxidation method was not able to provide 14C results, major difficulties were encountered for 41Ca (spectral interference and quenching) and 55Fe (incomplete dissolution of matrix and yield correction) analyses, and 63Ni analyses would have required minimum of 5 g subsamples and an LSC sample as colorless as possible, for obtaining results above limit of detection.
As a conclusion, all methods are potential starting points for analysing activated concrete, but further development is needed, e.g., for improving especially radiochemical yield of 41Ca and overcome quenching problems in LSC. The main problems originated from low solubility and low activity concentration of the activated concrete, then high sample mass was required for analysis, which in turn resulted in high amount of stable interfering elements. More results exceeding detection limit are needed in order to fully compare the methods with each other.
References
Hou X (2018) Liquid scintillation counting for determination of radionuclides in environmental and nuclear applications. J Radioanal Nucl Chem 318:1597–1628
Hou X (2005) Radiochemical determination of 41Ca in nuclear reactor concrete. Radiochim Acta 93:611–617
Warwick PE, Kim D, Croudace IW, Oh J (2010) Effective desorption of tritium from diverse solid matrices and its application to routine analysis of decommissioning materials. Anal Chim Acta 676:93–102
Kim JK, Warwick PE, Croudace IW (2008) Tritium speciation in nuclear reactor bioshield concrete and its impact on accurate analysis. Anal Chem 80:5476–5480
Dinali GS, Ramos SJ, de Carvalho TS, Carvalho GS, de Oliveira C, Siqueira JO, Guilherme LRG (2018) Dissolution techniques for determination of rare earth elements in phosphate products: acid digestion or alkaline fusion? J Geochem Explor 197:114–121
Ishimori K, Kameo Y, Matsue H, Ohki Y, Nakashima M, Takahashi K (2011) Carbon-14 analysis in solidified product of non-metallic solid waste by a combination of alkaline fusion and gaseous CO2 trapping. Appl Radiat Isot 69:506–510
Maxwell SL, Culligan BK, Kelsey-Wall A, Shaw PJ (2011) Rapid radiochemical method for determination of actinides in emergency concrete and brick samples. Anal Chim Acta 701:112–118
Delijska A, Blazheva T, Petkova L, Dimov L (1988) Fusion with lithium borate as sample preparation for ICP and AAS analysis. Fresenius Z Anal Chem 332:362–365
Braysher E, Russell B, Woods S, García-Miranda M, Ivanov P, Bouchard B, Read D (2019) Complete dissolution of solid matrices using automated borate fusion in support of nuclear decommissioning and production of reference materials. J Radioanal Nucl Chem 321:183–196
Banba T, Hagiya H, Tamura Y, Senoo M, Yonezawa C, Carter PB (1998) Chemical analysis of high-level radioactive waste glass by ICP-AES. Anal Sci 14:389–394
Rudisill TS, Karraker DG (2001) Development of a sodium peroxide pretreatment process for the recovery of plutonium from refractory residues. WSRC-TR-2001–00348, Westinghouse Savannah River Company, US Department of Energy, https://doi.org/10.2172/787828
Leskinen A, Salminen-Paatero S, Räty A, Tanhua-Tyrkkö M, Iso-Markku T, Puukko E (2020) Determination of 14C, 55Fe, 63Ni and gamma emitters in activated RPV steel samples—a comparison between calculations and experimental analysis. J Radioanal Nucl Chem 323:399–413
Leskinen A, Salminen-Paatero S, Gautier C, Räty A, Tanhua-Tyrkkö M, Fichet P, Kekki T, Zhang W, Bubendorff J, Laporte E, Lambrot G, Brennetot R (2020) Intercomparison exercise on difficult to measure radionuclides in activated steel: statistical analysis of radioanalytical results and activation calculations. J Radioanal Nucl Chem 324:1303–1316
Hazan I, Korkisch J (1965) Anion-exchange separation of iron, cobalt and nickel. Anal Chim Acta 32:46–51
Räty A, Kekki T, Tanhua-Tyrkkö M, Lavonen T, Myllykylä E (2018) Preliminary waste characterization measurements in FiR 1 TRIGA research reactor decommissioning project. Nucl Techn 203(2):205–220
Räty A, Lavonen T, Leskinen A, Likonen J, Postolache C, Fugaru V, Bubueanu G, Lungu C, Bucsa A (2019) Characterization measurements of fluental and graphite in FiR1 TRIGA research reactor decommissioning waste. Nucl Eng Des 353:110198
Räty A (2020) Activity characterisation studies in FiR1 TRIGA research reactor decommissioning project, Doctoral school in natural sciences dissertation series, URN:ISSN:2670-2010
Beckurts KH, Wirtz K (1964) Neutron physics, Appendix I 407–416. Springer, Berlin
Brennetot R, Giuliani M, Guegan S, Fichet P, Chiri L, Deloffre A, Mougel C, Bachelet F (2017) 3H measurement in radioactive wastes: efficiency of the pyrolysis method to extract tritium from aqueous effluent, oil and concrete. Fusion Sci Technol 71:397–402
Hou X (2005) Rapid analysis of 14C and 3H in graphite and concrete for decommissioning of nuclear reactor. Appl Radiat Isot 62:871–882
Hou X, Frosig Ostergaard L, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste samples using radiochemical separation and liquid scintillation counting. Anal Chim Acta 535:297–307
Eriksson S, Vesterlund A, Olsson M, Ramebäck H (2013) Reducing measurement uncertainty in 63Ni measurements in reactor coolant water high in 60Co activities. J Radioanal Nucl Chem 296:775–779
Eichrom method (2014a) Iron-55 in water, No. FEW01, revision 1.1, Eichrom Technologies LLC
Ervanne H, Hakanen M, Lehto J, Kvarnström R, Eurajoki T (2009) Determination of 45Ca and γ-emitting radionuclides in concrete from a nuclear power plant. Radiochim Acta 97:631–636
Itoh M, Watanabe K, Hatakeyama M, Tachibana M (2002) Determination of 41Ca in biological-shield concrete by low-energy X-ray spectrometry. Anal Bioanal Chem 372:532–536
Nottoli E, Bourles D, Bienvenu P, Labet A, Arnold M, Bertaux M (2013) Accurate determination of 41Ca concentrations in spent resins from the nuclear industry by accelerator Mass spectrometry. Appl Rad Isot 82:340–346
Warwick P, Croudace I (2006) Isolation and quantification of 55Fe and 63Ni in reactor effluents using extraction chromatography and liquid scintillation analysis. Anal Chim Acta 567:277–285
Eichrom Method (2014b) Nickel-63/59 in water, No. NIW01 analytical procedure, revision 1.3, Eichrom Technologies LLC
Lee C, Martin JE, Griffin HC (1997) Optimization of measurement of 63Ni in reactor waste samples using 65Ni as a tracer. Appl Radiat Isot 48:639–642
Currie LA (1968) Limits for qualitative detection and quantitative determination. Appl Radiochem Anal Chem 40(3):586–593
Säteilyturvakeskuksen määräys vapaarajoista ja vapauttamisrajoista [2018] Määräys STUK SY/1/2018
Hou X (2007) Radiochemical analysis of radionuclides difficult to measure for waste characterization in decommissioning of nuclear facilities. J Radioanal Nucl Chem 273:43–48
Leskinen A, Tanhua-Tyrkkö M, Salminen Paatero S, Laurila J, Kurhela K, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Laporte E, Guiliani M, Bubendorff J, Fichet P (2021). DTM-Decom II - Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-441, NKS-B, Roskilde, Denmark
Leskinen A, Gautier C, Räty A, Kekki T, Laporte E, Giuliani M, Bubendorff J, Laurila J, Kurhela K, Pascal F, Salminen-Paatero S (2021) Intercomparison exercise on difficult to measure radionuclides in activated concrete—statistical analysis and comparison with activation calculations. J Radioanal Nucl Chem 329:945–958
Acknowledgements
The research was funded by KYT 2022 (Finnish Research Programme on Nuclear Waste Management 2019-2022). The authors would also like to thank VTT personnel and FiR1 decommissioning task force for their collaborative actions and provision of the studied material.
Funding
Open Access funding provided by Technical Research Centre of Finland (VTT).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leskinen, A., Salminen-Paatero, S. Development of 3H, 14C, 41Ca, 55Fe, 63Ni radiochemical analysis methods in activated concrete samples. J Radioanal Nucl Chem 331, 31–41 (2022). https://doi.org/10.1007/s10967-021-08073-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-021-08073-4