
Abstract
Due to its favorable decay properties, the non-standard radionuclide 45Ti is a promising PET isotope for tumor imaging. Additionally, titanium complexes are widely used as anti-tumor agents and 45Ti could be used to study their in vivo distribution and metabolic fate. However, although 45Ti can be obtained using the 45Sc(p,n)45Ti nuclear reaction its facile production is offset by the high oxophilicity and hydrolytic instability of Ti4+ ions in aqueous solutions, which complicate recovery from the irradiated Sc matrix. Most available 45Ti recovery procedures rely on ion exchange chromatography or solvent extraction techniques which are time-consuming, produce large final elution volumes, or, in case of solvent extraction, cannot easily be automated. Thus a more widespread application of 45Ti for PET imaging has been hampered. Here, we describe a novel, solvent-free approach for recovery of 45Ti that involves formation of [45Ti]TiCl4 by heating of an irradiated Sc target in a gas stream of chlorine, followed by thermochromatographic separation of the volatile radiometal chloride from co-produced scandium chloride and trapping of [45Ti]TiCl4 in a glass vial at − 78 °C. The recovery of 45Ti amounted to 76 ± 5% (n = 5) and the radionuclidic purity was determined to be > 99%. After trapping, the [45Ti]TiCl4 could be directly used for 45Ti-radiolabeling, as demonstrated by the successful radiosynthesis of [45Ti][Ti(2,4-salan)].
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Discovery of the anticancer activity of cisplatin and its clinical introduction in the 1970s have spurred interest into metal based antitumor compounds with less side effects and increased effectiveness against a broad range of cancers [1, 2]. Titanium(IV) complexes like budotitane, titanocene dichloride and their derivatives are effective against various cancer cell lines but failed in in vivo clinical trials [3,4,5,6], most likely due to their rapid (within seconds) hydrolysis under physiological conditions [7]. A more promising class of titanium-based drugs with hydrolytic half-lives in the range of hours is based on tetradentate diaminobis(phenolato) ligands (salans) [8]. Their titanium complexes selectively induce apoptotic cell death [9] and display strong antitumor properties in vitro and in tumor-bearing mice [10, 11]. Further studies on their distribution, uptake and mechanism of action rely on imaging techniques such as positron emission tomography (PET), which allows for non-invasive assessment of the biological fate of radiolabeled drugs while they distribute in vivo. The titanium isotope 45Ti has a half-life of 3.1 h, a high positron branching ratio (β+ = 84.8%) and low maximum positron energy (Eβ+max = 439 keV), negligible secondary gamma emission and a low β end point energy of 1.04 MeV, making it an ideal candidate for use in PET studies [12,13,14]. However, while the radiometal can be produced by transmutation of naturally monoisotopic scandium with low energy protons [15, 16], 45Ti radiochemistry is hampered by the high oxophilicity (θ = 1.0) and hydrolytic instability of Ti4+ ions in aqueous environments, which necessitate the use of strongly acidic conditions [17, 18]. Solid phase extraction of 45Ti dissolved in acidic solutions by ion exchange chromatography is time-consuming, cannot easily be automated and often results in non-reactive titanyl species that are unsuitable for production of titanium complexes [19, 20]. A number of approaches have been proposed to circumvent these problems, which include the use of hydroxamate resins and oxalic acid elution [21], trapping of 45Ti on a diol-functionalized resin followed by on-resin radiosynthesis [22] and continuous liquid–liquid extraction of the radioisotope with a guaiacol/anisole mixture to obtain a 45Ti containing organic phase that can be used for radiolabeling [23, 24]. Here, we describe an alternative, solvent-free “one-pot” method that is based on thermochromatographic separation of 45Ti from an irradiated Sc target and consecutive radiotitanation of a salan ligand, thereby obviating the need for organic solvents, solid phase extraction or on-column chelation chemistry.
Experimental
Materials
Dry tetrahydrofurane (THF), N,N-diisopropylethylamine (DIPEA) (99.5%), 2,4-dimethylphenol (98%) and thin layer chromatography (TLC) plates were purchased from Sigma-Aldrich (Germany). N,N′-bis(2-hydroxyethyl)ethylendiamine (95%) was purchased from ABCR GmbH (Germany). Scandium ingots were purchased from Smart Elements GmbH (Austria). Chlorine gas (5.0) was obtained from Linde Gas (Germany). All chemicals were used without further purification. Mass flow controllers were bought from Bronkhorst Deutschland Nord GmbH (Germany) (EL-FLOW Select 300 mL/min for inert gases, LOW-ΔP-FLOW 60 mL/min for chlorine gas). Mass flow conversions were done using the Fluidat database (Bronkhorst Nord GmbH, Germany). Glassware for the separation system was manufactured at the Central Institute of Engineering, Electronics and Analytics (ZEA-1) at Forschungszentrum Jülich.
Analytical instrumentation
Gamma spectroscopy was performed with ORTEC gamma-ray spectrometers (AMETEK GmbH, Germany), which were energy and efficiency calibrated with certified radiation point sources (Co-60, Ba-133, Eu-152, Ra-226) from the Physikalisch-Technische Bundesanstalt (Germany). To monitor the separation process, a Geiger–Müller–Tube (VacuTec GmbH, Germany) was used. Radio-TLC measurements were performed on 60F254 silica plates (Merck, Germany) with methanol as mobile phase and recorded using a Packard Instant Imager (Packard Instrument Company, USA). The radioactivity detection limit (LOD) was determined by serial dilution and amounted to ≤ 3 Bq for an exposure time of 30 min. Radio-HPLC was performed on an Azura P 4.1 s pump with an Azura UVD 2.1 s UV/VIS detector (Knauer Wissenschaftliche Geräte GmbH, Germany) and an EG & G Ortec ACE NaI(Tl) radioactivity detector with photomultiplier (EG & G Ortec, USA). The radioactivity detection limit was determined by serial dilution and amounted to 0.3 kBq. Co-elution experiments of radioactive and non-radioactive complexes were performed with a flow rate of 1 mL/min using H2O/MeOH/CH3COOH (27/73/0.07, v/v/v) as the mobile and Synergi Fusion 4 µ RP 80 Å, 250 × 4.6 mm (Phenomenex Inc., Germany) as the RP stationary phase. The UV detection wavelength for all measurements was 240 nm. NMR spectra were measured on a Varian Inova 400 spectrometer (Agilent Technologies, Germany) with 400.1 MHz (1H-NMR) and 100.62 MHz (13C-NMR). High-resolution mass spectrometry (HRMS) was performed using a hybrid linear ion trap FT-ICR mass spectrometer LTQ-FT Ultra (Thermo Fisher Scientific, Germany) equipped with a 7 T superconducting magnet. The electrospray ionisation (ESI) source was operated in the positive mode. Low-resolution mass spectrometry was measured using a Finnigan Automass Multi spectrometer (Thermoquest, Germany). 45Sc concentrations in solution were determined by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 7500 instrument with quadrupole mass analyzer and collision cell (Agilent Technologies, Germany). The instrument was operated with helium as collision gas to minimize spectral interferences by cluster ions. Quantification was performed by external calibration using Rh as the internal standard.
Radionuclide production
Titanium-45 was produced from discs of metallic scandium (0.35 ± 0.1 g) by the 45Sc(p,n)45Ti nuclear reaction. As target material scandium of 99.99% purity was used (Smart-Elements GmbH, Austria). The material came in form of small ingots, which were rolled to plates of 0.6 mm. From those plates circular target of 13 mm were cut and put into a copper target holder to be irradiated at the solid target station of the BC1710 [30]. The target was irradiated with 8.2–16.9 MeV protons at 1.5 µA for 30 min using the Baby Cyclotron BC1710 at the INM-5 (Forschungszentrum Jülich). To minimize coproduction of 44Sc (half-life: 3.9 h), 44mSc (half-life: 58 h) and 44Ti (half-life: 60 y), two Ni foils with a thickness of 125 µm each were used to degrade the proton energy to approximately 12 MeV. For initial optimization studies, irradiation was performed without degradation of the proton beam, so that 44Sc and 44mSc could be used to radiometrically monitor the separation process and determine the radiochemical purity of the product.
Radionuclide separation system
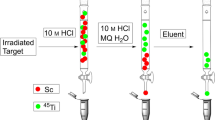
The separation set-up (Fig. 1) consisted of a quartz glass reaction chamber that was connected to the main gas line via a Young valve and could be heated to temperatures of up to 1000 °C by a model MTF 12/39/250 heating oven (Carbolite Gero GmbH, Germany), a borosilicate glass tube condenser that could be heated to 400 °C, a receiving flask that could be cooled down to − 79 °C by an acetone/CO2 cooling bath and a chlorine scrubbing system with 20% sodium hydroxide solution. Various gas mixtures were supplied to the setup by two mass flow controllers (Fig. 2).

Schematic representation of the separation setup

Scheme of the mass-flow-controller setup
The ground glass joint connecting the reaction chamber and the condenser was sealed with a polytetrafluoroethylene sealing ring to obviate the need for high vacuum grease which could have absorbed or contaminated the product. In addition, the upper part of the receiving flask was equipped with a septum, which allowed for addition of reagents during the radiosynthesis that was performed immediately after the separation process.
Thermochromatographic separation of 45Ti from scandium
For isolation of titanium-45, the irradiated Sc matrix was placed into the reaction chamber, which was flushed with helium (125 mL/min) and heated to 900 °C. When the reaction temperature was reached, the chamber was flushed with a mixture of chlorine (15 mL/min) and helium (110 mL/min) gas to form the halides [45Ti]TiCl4 and ScCl3 respectively. As the gaseous mixture of metal chlorides reached the condenser, ScCl3 (boiling point ~ 975 °C) resublimed while [45Ti]TiCl4 (boiling point ~ 136 °C) passed further and condensed in the receiving flask. A Geiger–Müller counter was used to monitor the separation process. When the collection of [45Ti]TiCl4 was complete, the addition of chlorine and heating were discontinued, the temperature of the receiving flask was raised to − 30 °C and the system was flushed with helium for 10 min to remove excess chlorine. Optimal reaction conditions (temperature, chlorine concentration and gas flow) were determined experimentally as described in more detail in the results section and supporting information.
Synthesis of 6,6′-((ethane-1,2-diylbis((2-hydroxyethyl)azanediyl))bis(methylene))bis(2,4-dimethyl-phenol) (2,4-salanH4)
740 mg (1 eq, 5.0 mmol) 1,2-bis(2-aminoethoxy)ethane, 122 mg 2,4-dimethylphenol (2 eq, 10.0 mmol) and 500 mg paraformaldehyde (3.3 eq, 16.7 mmol) were dissolved in methanol and refluxed for 12 h. The organic solvent was removed under reduced pressure and the crude product recrystallized from diethylether/petrolether to obtain the target compound as white crystals. (251 mg, yield: 12%). 1H-NMR (400 MHz, CDCl3): δ 6.89 (br, 2 h, Ar), 6.65 (s, 2 h, Ar), 3.82–3.76 (m, 2 h, CH2), 3.71 (s, 4 h, CH2), 2.75 (s, 4 h, CH2), 2.23 (s, 6 h, CH3), 2.22 ppm (s, 6 h, CH3). 13C-NMR (101 MHz, CDCl3): 152.3, 131.0, 128.3, 127.3, 125.0, 121.0, 58.9, 57.2, 54.8, 50.8, 20.4, 15.7 ppm. HRMS: m/z calculated for [M + H]+ C24H37N2O4: 417.27533, found: 417.27481 [M + H]+.
Synthesis of titanium complex [Ti(2,4-salan)]
Under argon atmosphere, 150 mg 6,6′-((ethane-1,2-diylbis((2-hydroxyethyl)azanediyl))bis-(methylene))-bis(2,4-dimethylphenol) (2,4-salanH4) was dissolved in 25 ml dry THF and 139 mg titanium isopropoxide was added to the solution. After stirring for 15 min the organic solvent was removed under reduced pressure and the product was obtained as light yellow solid (220 mg, yield: 99%). 1H-NMR (400 MHz [D6]DMSO): δ 6,86 (s, 2 h, Ar), 6,78 (s, 2 h, Ar), 4,45–4,25 (m, 4 h, CH2), 3,79 (br, J = 18,0 4 h, CH2), 3,51 (d, J = 8,6 Hz, 2 h, CH2), 3,23 (br, 2 h, CH2), 3,12 (d, J = 8,9 Hz, 2 h, CH2), 2,85 (d, J = 9,5 Hz, 2 h, CH2), 2,18 (s, 6 h, CH3), 2,12 (s, 6 h, CH3). HRMS: m/z calculated for [M + H]+ C24H33O4N2Ti: 461.19198, found: 461.19148 [M + H]+.
Radiosynthesis of [45Ti][Ti(2,4-salan)]
2 mg 2,4-salanH4 and 20 µl of DIPEA in 2 mL anhydrous THF were added to the receiving flask containing the isolated [45Ti]TiCl4 and allowed to react for 5 min at − 30 °C, after which the mixture was analyzed by HPLC. For purification of the radiolabeled complex, the reaction mixture was diluted with H2O (30 mL) and loaded onto a SepPak C18 RP cartridge (Waters GmbH, Germany). The cartridge was washed with a mixture of H2O/MeOH/CH3COOH (4/1/0.005 mL) and the product eluted with MeOH (3 mL).
Results and discussion
Routine production of 45Ti by proton bombardment of natural scandium resulted in high radionuclidic purity and good yields. The latter amounted to about 290 MBq for a typical bombardment with 12 MeV protons and 1.5 µA current for 30 min of beam time. When choosing 16.9 MeV incident proton energy, a typical bombardment of 1.5 µA for 30 min produced a batch yield of 360 MBq and the product was composed of 87% 45Ti, 12% 44Sc and 1% 44mSc. These production yields are comparable to those reported by Vavere et al. [19]. For isolation of the radiometal and formation of radiolabeled complexes, we developed a solvent-free “one-pot” procedure that involves (1) formation of [45Ti]TiCl4 by reaction of the cyclotron-irradiated scandium target with chlorine gas, (2) isolation of [45Ti]TiCl4 by thermochromatographic separation of the resulting metal chlorides and (3) direct reaction of isolated [45Ti]TiCl4 with a chelating ligand to form the radiometal complex.
Preparation of [45Ti]TiCl4 by high-temperature chlorination of irradiated Sc
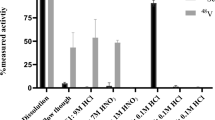
To avoid the formation of titanyl species, [45Ti]TiCl4 for PET radiochemistry has almost exclusively been produced by dissolving irradiated scandium targets with concentrated hydrochloric acid. Recovery of the radiometal chloride from acidic solutions involves arduous separation chemistry, which has limited a more widespread application of 45Ti for PET imaging. To circumvent these problems, we investigated high-temperature chlorination, which has long been used for commercial TiCl4 production from rutile (TiO2) or ilmenite (FeO–TiO2) feedstocks [25]. To this end, we first examined whether [45Ti]TiCl4 could be produced and separated from the irradiated target under conditions that minimize formation of scandium chlorides. However, preliminary experiments with non-irradiated targets treated with 100% Cl2 demonstrated that formation of ScCl3 starts to occur at temperatures above 600 °C which is still below the 850–1050 °C typically used for chlorination of titanium ores [25]. Also, no formation of [45Ti]TiCl4 was observed when an irradiated target was chlorinated at 550 °C. When the temperature was increased to 750 °C, some formation of [45Ti]TiCl4 took place but isolation was hampered by an exothermic reaction of the scandium with Cl2, which contaminated the whole separation apparatus. To avoid this exothermic reaction, a series of optimization experiments was performed with a fixed reaction temperature of 900 °C and a total gas flow rate of 100 mL/min while varying the concentration of chlorine. For these experiments, irradiation of the Sc target was deliberately performed without degradation of the proton beam, so that 44Sc and 44mSc could be used to radiometrically monitor the separation. As illustrated in Table 1, 45Ti recovery under these conditions was maximal (53%) when the chlorine concentration was 12%. In a second set of optimization experiments, temperature and chlorine concentration were fixed at 900 °C and 12% while the total gas flow rate was varied (Table 2). The results showed that highest recovery rates could be obtained with total gas flow rates between 100 and 150 mL/min, whereas recovery decreased at lower or higher rates (Table 2). Finally, we examined the influence of reaction temperature on recovery of 45Ti with the total flow rate fixed to 125 mL/min and a Cl2 concentration of 12%. The recovery of [45Ti]TiCl4 decreased as the temperature was lowered to 850 °C (Table 3), presumably because ScCl3 formation on the surface of the target prevented reaction of the radiometal with Cl2. Better results (53% recovery) were obtained at 900 °C, a temperature at which sublimation of ScCl3 was sufficient to prevent its accumulation on the surface of the target. If the temperature was further increased to 1000 °C, the recovery yield decreased again by 13%, most likely reflecting the thermal decomposition of [45Ti]TiCl4 at that temperature [26, 27].
Thermochromatographic separation and purification of [45Ti]TiCl4
As noted above and consistent with previous reports [28, 29], appreciable transition of ScCl3 to the gas phase was observed at temperatures above 850 °C, requiring its removal from the gaseous mixture of metal chlorides. This was initially attempted with fritted glass, which proved to have several disadvantages. Apart from loss of product on the large surface area of the frit, re-sublimation of ScCl3 tended to clog the system, which could potentially result in dangerous pressure build-up. Therefore, a simple condenser made of a 100 cm borosilicate spiral coil that was heated to 400 °C and served as the vapor–condensate path was used instead. In initial experiments with argon as the inert gas, no separation could be achieved since ScCl3 was transported to the receiving flask. In contrast using helium as the inert gas, ScCl3 resublimed in the condenser and was effectively retained, while [45Ti]TiCl4 passed through and could be collected in the receiving flask. A temperature of − 78° was chosen to condense the radiometal chloride (boiling point 136 °C) while avoiding formation of solid Cl2 (melting point − 101.5 °C) in the receiving flask. Once the collection of [45Ti]TiCl4 was complete, remaining Cl2 (boiling point − 34.04 °C) could be easily removed by increasing the temperature to − 30 °C and flushing the trapping vessel with helium gas for 10 min. Tracer experiments with 44Sc and gamma-ray spectroscopy showed that negligible 44,44mSc activities were found in the isolated 45Ti fraction. Additionally, ICP-MS measurements of Sc in the product fraction confirmed that only traces of Sc (0.005 mg (0.1 ppm)) were transported to the trapping vessel.
Finally, the optimized reaction conditions were used for the separation of pure 45Ti, produced by degrading the proton energy for irradiation of the Sc target to approximately 12 MeV (see Experimental section). This allowed precise quantification of 45Ti recovery by simply dividing the recovered activity of 45Ti by the total starting activity. On average, recovery of 45Ti amounted to 76 ± 5% (n = 5). The separation process was conducted within 2 h, corresponding to a non-decay-corrected (n.d.c.) recovery of 48 ± 3% (n = 5) (Table 4). The amount of non-radioactive Ti was analyzed by ICP-MS to be less than 1 µg/L in the final reaction volume.
Radiosynthesis of [45Ti][Ti (2,4-salan)]
To confirm that the isolated [45Ti]TiCl4 can be directly employed for radiolabeling, it was reacted with the hexadentate ligand 2,4-salan to produce a [45Ti][Ti(2,4-salan)] complex (Fig. 3). To this end, 2,4-salan and DIPEA were dissolved in dry THF, added to the receiving flask containing the purified [45Ti]TiCl4 and allowed to react for 5 min at − 30 °C. HPLC of the reaction mixture with co-injection of the non-radioactive reference compound confirmed the identity of the product and the absence of radioactive byproducts (Fig. 4). However, radio-TLC revealed up to 50% of an unidentified byproduct, presumably [45Ti]TiO2, which may have formed due to traces of H2O present in the reaction mixture. From the starting activity used for this reaction, 50% could be recovered from the receiving vessel for further purification. The radiochemical yield (RCY) obtained after isolation of the radiocomplex via solid phase extraction amounted to 15 ± 7% (n = 7).

Formation of [45Ti][Ti(2,4-salan)] complex by radiolabeling of 2,4-salanH4 with [45Ti]TiCl4 and DIPEA

HPLC chromatogram of [45Ti][Ti(2,4-salan)] with coinjected reference compound. Mobile phase: H2O/MeOH/CH3COOH, 27/73/0.07, vol/vol/vol. Stationary phase: Synergy Fusion RP, flow = 1 mL/min. The UV-peaks at ≈ 4 min correspond to protonated excess ligand and base
Conclusions
In this work a radioseparation method of no-carrier-added [45Ti]TiCl4 from Sc is described. 45Ti was produced by proton irradiation of a metallic Sc target followed by thermochromatographic separation in a chlorine gas stream. The separated [45Ti]TiCl4 could be used directly for the preparation of the radiometal complex [45Ti][Ti(2,4-salan)] in radiochemical yields of 10–20%. This simple separation technique obviates the need for complex separation chemistry and may contribute to a more widespread application of 45Ti-labeled compounds in imaging applications.
References
Gaynor D, Griffith DM (2012) The prevalence of metal-based drugs as therapeutic or diagnostic agents: beyond platinum. Dalton Trans 41:13239–13257
Mjos KD, Orvig C (2014) Metallodrugs in medicinal inorganic chemistry. Chem Rev 114:4540–4563
Schilling T, Keppler KB, Heim ME, Niebch G, Dietzfelbinger H, Rastetter J, Hanauske A-R (1995) Clinical phase I and pharmacokinetic trial of the new titanium complex budotitane. Invest New Drugs 13:327–332
Lümmen G, Sperling H, Luboldt H, Otto T, Rübben H (1998) Phase II trial of titanocene dichloride in advanced renal-cell carcinoma. Cancer Chemother Pharmacol 42:415–417
Strohfeldt K, Tacke M (2008) Bioorganometallic fulvene-derived titanocene anti-cancer drugs. Chem Soc Rev 37:1174–1187
Meléndez E (2002) Titanium complexes in cancer treatment. Crit Rev Oncol Hematol 42:309–315
Toney JH, Marks TJ (1985) Hydrolysis chemistry of the metallocene dichlorides M(.eta.5-C5H5)2Cl2, M = titanium, vanadium, or zirconium. Aqueous kinetics, equilibria, and mechanistic implications for a new class of antitumor agents. J Am Chem Soc 107:947–953
Immel TA, Groth U, Huhn T (2010) Cytotoxic titanium salan complexes: surprising interaction of salan and alkoxy ligands. Chem A Eur J 16:2775–2289
Immel TA, Debiak M, Groth U, Bürkle A, Huhn T (2009) Highly selective apoptotic cell death induced by halo-salane titanium complexes. ChemMedChem 4:738–741
Immel TA, Groth U, Huhn T, Öhlschläger P (2011) Titanium salan complexes displays strong antitumor properties in vitro and in vivo in mice. PLoS ONE 6:e17869
Immel TA, Grützke M, Späte A-K, Groth U, Öhlschläger P, Huhn T (2012) Synthesis and X-ray structure analysis of a heptacoordinate titanium(iv)-bis-chelate with enhanced in vivo antitumor efficacy. Chem Commun 48:5790–5792
Kuhn S, Spahn I, Scholten B, Coenen HH (2015) Positron and γ-ray intensities in the decay of 45Ti. Radiochim Acta 103:403–409
Burrows TW (2008) Nuclear data sheets for A = 45. Nucl Data Sheets 109:171–296
Chaple LF, Lapi SE (2018) Production and use of the first-row transition metal PET radionuclides 43,44Sc, 52Mn, and 45Ti. J Nucl Med 59:1655–1659
Sadeghi M, Enferadi M, Nadi H (2011) 45Ti, a candidate for positron emission tomography: study of the cyclotron production. Radiochemistry 53:411–414
Fazaeli Y, Aboudzadeh M, Aardaneh K, Kakavand T, Bayat F, Yousefi K (2014) A new approach to targetry and cyclotron production of 45Ti by proton irradiation of 45Sc. Nucl Technol Radiat Prot 29:28–33
Kepp KP (2016) A quantitative scale of oxophilicity and thiophilicity. Inorg Chem 55:9461–9470
Kislik V (2002) Competetive complexation/solvation theory of solvent extraction. II. Solvent extraction of metals by acidic extractants. Sep Sci Technol 37:2623–2657
Vāvere AL, Laforest R, Welch MJ (2005) Production, processing and small animal PET imaging of titanium-45. Nucl Med Biol 32:117–122
Kislik V, Eyal A (1993) Acidity dependence of Ti(IV) extraction: a critical analysis. Solvent Extr Ion Exch 11:258
Gagnon K, Severin GW, Barnhart TE, Engle JW, Valdovinos HF, Nickles RJ (2012) 45Ti extraction using hydroxamate resin. In: AIP conference proceedings , vol 1509, p 211
Severin GW, Nielsen CH, Jensen AI, Fonslet J, Kjær A, Zhuravlev F (2015) Bringing radiotracing to titanium-based antineoplastics: solid phase radiosynthesis, PET and ex vivo evaluation of antitumor agent [45Ti](salan)Ti(dipic). J Med Chem 58:7591–7595
Pedersen KS, Imbrogno J, Fonslet J, Lusardi M, Jensen KF, Zhuravlev F (2018) Liquid–liquid extraction in flow of the radioisotope titanium-45 for positron emission tomography applications. React Chem Eng 3:898–904
Søborg Pedersen K, Baun C, Michaelsen Nielsen K, Thisgaard H, Ingemann Jensen A, Zhuravlev F (2020) Design, synthesis, computational, and preclinical evaluation of natTi/45Ti-labeled urea-based glutamate PSMA ligand. Molecules 25:1104
Bordbar H, Yousefi AA, Abedini H (2017) Production of titanium tetrachloride (TiCl4) from titanium ores: a review. Polyolefins J 4:149–173
Teyssandier F, Allendorf MD (1998) Thermodynamics and kinetics of gas-phase reactions in the Ti–Cl–H system. J Electrochem Soc 145:2167–2178
Herzler J, Roth P (2002) High-temperature decomposition of TiCl4 based on Cl-concentration measurements. Proc Combust Inst 29:1353–1359
Biltz W, Klemm W (1923) Über die elektrolytische Leitfähigkeit geschmolzenen Scandiumchlorids. Z für Anorg Allg Chem 131:22–26
Voigt A, Biltz W (1924) Über das elektrolytische Leitvermögen geschmolzener chloride. Z für Anorg Allg Chem 133:277–305
Spellerberg S, Scholten B, Spahn I, Bolten W, Holzgreve M, Coenen HH, Qaim SM (2015) Target development for diversified irradiations at a medical cyclotron. Appl Radiat Isot 104:106–112
Acknowledgements
The authors are grateful to Mr. S. Spellerberg for technical assistance, and to Mr. M. Holzgreve and Mr. I. Montag for all cyclotron-irradiations. Further thanks go to F. Neumaier for his assistance in the preparation of this manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interests is reported.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Giesen, K., Spahn, I. & Neumaier, B. Thermochromatographic separation of 45Ti and subsequent radiosynthesis of [45Ti]salan. J Radioanal Nucl Chem 326, 1281–1287 (2020). https://doi.org/10.1007/s10967-020-07376-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07376-2