
Abstract
The totally symmetric stretching mode \(\nu_{1}\) Ln–(OH2) of the first hydration shells of all the rare earth (RE) ions across the series from lanthanum to lutetium has been measured on dilute aqueous perchlorate solutions at room temperature. An S-shaped relationship has been found between the \(\nu_{1}\) Ln–(OH2) peak positions and the Ln–(OH2) bond distances of the lanthanide(III) aqua ions. While the light rare earth ions form nona-hydrates, the heavy ones form octa-hydrates and the rare earth ions in the middle of the series show non integer hydration numbers between 9 and 8. A relationship between wavenumber positions \(\nu_{1}\) Ln–(OH2) and the Ln–(OH2) bond distances of the RE hydrates has been given. Recent quantum mechanical calculations support the given interpretation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The trivalent lanthanides (Ln3+) are part of the longest series of chemically similar metal ions marked by progressive filling of the 4f orbitals going from La3+ to Lu3+. A subsequent decrease of the ionic radii along the series is observed due to the increase of the nuclear electric field. The decrease of the ionic radii of the rare earth ions with increasing atomic number, known as lanthanide contraction [1, 2], was first coined by Goldschmidt [2]. A smooth plot of thermodynamic properties of lanthanide salts in aqueous solution would, therefore, be expected. This is especially true because the f-electrons do not take part in the bonding (negligible ligand field effect).
Various physical and chemical properties of the series of rare earth ions in aqueous solution have been studied over the years (apparent molal volumes, relative viscosities, apparent molal heat capacities, heats of dilution and electrical conductance [3,4,5,6]). However, many thermodynamic and transport properties of rare earth ions in aqueous solution actually follow an irregular or S-shaped behavior when plotted against the atomic number or the ionic radius [3,4,5,6,7,8,9]. This behavior was seen as the result of the coordination number change of the first sphere in the middle of the lanthanide series following neodymium to terbium (see [7] and references therein). From X-ray and neutron diffraction measurements [10,11,12,13,14,15,16,17], it was deduced that the light rare earth ions exist as nona-hydrates, the heavy ones octhydrates and the ions in the middle of the series are mixtures of both (see ref [7].). Recent extended X-ray absorption fine structure (EXAFS) techniques confirmed these coordination structures [18, 19]. Theoretical simulations [20,21,22,23] on the rare earth ion hydrates have also been carried out and supported the picture given by experimental structure techniques.
The tetrad effect [24] and the gadolinium break (half-filled f electron shell) [25] leads to small deviations from the expected smooth curvature. These effects are caused by the quantum mechanical inter electronic repulsion energy of the q electrons in the 4 fq electronic configuration [26]. The tetrad effect is much smaller than the effect caused by the change in coordination number across the whole lanthanide(III) series and it should be mentioned that the tetrad effect is not always observable [27].
In recent years, we characterized the hydrates of light rare earth ions and the members of the heavy ones in aqueous solution using Raman spectroscopy [28,29,30]. Raman spectroscopic data in conjunction with Ln–O bond distances are helpful to further shed light on the structure of the rare earth ions. If the totally symmetric stretching modes of these ions, \(\nu_{1}\) Ln–(OH2) were to follow a rise with atomic number or decreasing ionic radius of these ions (lanthanide contraction) then the symmetric stretching modes \(\nu_{1}\) Ln–OH2 would monotonically increase.
As an extension of our previous work on rare earth ions in solution, the Raman spectra were measured in the middle of the series of the lanthanide(III) hydrates in order to complete the data for the whole series. The Raman spectra of perchlorate solutions of all rare earth ions, Ln(ClO4)3(aq), except promethium, are presented and discussed in this paper. Different concentrations of these rare earth ion solutions were measured, including fairly dilute solutions because the \(\nu_{1}\) Ln–(OH2) modes showed slight changes with concentration [28,29,30]. Therefore it is crucial to consider only dilute solutions when comparing the \(\nu_{1}\) Ln–(OH2) modes. Viewing our earlier results [28,29,30], it is apparent that the totally symmetric stretching modes,\(\nu_{1}\) Ln–(OH2) for these rare earth ions, plot on two different straight lines with respect to the atomic number z, or the Ln–O bond distances. The light rare earth ions forming nona-hydrates in aqueous solution plot on an almost straight line and the one for the heavy rare earth octa-hydrates on another. These spectroscopic results, including the ones for the trivalent rare earth (RE) ions in the middle of the series, will be discussed in detail, taking into account recent results concerning thermodynamic and transport properties, structural results as well as quantum mechanical calculations on these lanthanide hydrates.
2 Experimental Section
The rare earth perchlorate solutions of Eu(ClO4)3, Gd(ClO4)3, Tb(ClO4)3 and Dy(ClO4)3 were produced from their oxides which were dissolved with perchloric acid. The rare earth oxides were purchased from Sigma–Aldrich (now Merck, Darmstadt, Germany) at 99.9% purity on trace metal basis, except Tb2O3 with a purity of 99.99%. The HClO4 used was purchased from Merck, Darmstadt at 70% with p.a. quality. Ln(ClO4)3 stock solutions were prepared, from which more dilute solutions were prepared by weight with triply distilled water. The rare earth ion content of the individual solutions were determined by complexometric titration with a relative standard uncertainty of the complexometric titration is 1 × 10–3 [31]. The following solutions were prepared: Eu(ClO4)3(aq) at 1.110 and 0.370 mol·L–1; Gd(ClO4)3(aq) at 2.418 mol·L–1, 1.209 mol·L–1 and 0.403 mol·L–1; Tb(ClO4)3(aq) at 2.215 mol·L–1, 1.108 mol·L–1 and 0.316 mol·L–1; Dy(ClO4)3(aq): 2.554 mol·L–1, 1.277 mol·L–1 and 0.364 mol·L–1. Further details regarding solution preparations of Ln(ClO4)3(aq) and analytical procedures are given in previously published works [28,29,30].
Raman spectroscopic measurements have been described on many occasions in detail and therefore only a brief discussion may be given. The scattering geometries, excitation conditions and Raman equipment were the same as reported in earlier publications and for details see [32, 33]. Briefly, Raman spectra were measured in the macro chamber of the T 64,000 Raman spectrometer from Jobin Yvon in a 90° scattering geometry at 22 °C. A quartz cuvette was used (Hellma Analytics, Müllheim, Germany) with a 10 mm path length and a volume at 1000 µL. The spectra were excited with the 487.987 nm or the 514.532 nm line of an Ar+ laser at a power level of 1000 mW at the sample. The Eu(ClO4)3, Tb(ClO4)3 and Dy(ClO4)3 solutions were measured with the 514.532 nm Ar+ line while Gd(ClO4)3 solutions with the one at 487.987 nm. After passing the spectrometer in subtractive mode, with gratings of 1800 grooves/mm, the scattered light was detected with a cooled CCD detector. The scattering geometries IVV = (X[ZZ]Y) and IVH = (X[ZX]Y) are defined as follows: the propagation (wave vector direction) of the exciting laser beam is in X direction and the propagation of the observed scattered light is in Y direction, the 90° geometry. The polarization (electrical field vector) of the laser beam is fixed in the Z direction (vertical) and the observed scattered light is polarized in he Z direction (vertical) for the IVV scattering geometry and polarized in X direction (horizontal) for the IVH scattering geometry. Thus, for the two scattering geometries it follows:
and
The symbols \(\overline{\alpha }^{\prime}\) and \(\gamma^{\prime}\) denote the isotropic and the anisotropic invariant of the Raman polarizability tensor [31]. The isotropic spectrum, Iiso was constructed according to Eq. 3:
The polarization degree of the Raman bands, ρ (ρ = IVH/IVV) was determined using an analyzer and adjusted, if necessary, before each measuring cycle using CCl4 [32, 33].
In order to characterize the spectral features in the low wavenumber region, the Raman spectra in I-format were reduced and the R-spectra obtained in order to obtain spectra defined as \(R(\tilde{\nu })\) which are independent of the excitation wavenumber, \(\tilde{\nu }_{{\text{L}}}\); the measured Stokes intensity should be corrected for the scattering factor \((\tilde{\nu }_{{\text{L}}} - \tilde{\nu })^{3}\) in the case of applying counting methods [32, 33]. The spectra were further corrected for the Bose–Einstein temperature factor, \(B = [1 - \exp ( - h\tilde{\nu }c/kT)]\) and the frequency factor, \(\tilde{\nu }\), to give the so-called reduced spectrum, \(R(\tilde{\nu })\) and again details were given in earlier publications [32, 33]. The isotropic spectrum in R-format follows from the corrected RVV and RVH spectra in the same manner as those in I-format (see Eq. 3). In the low wavenumber region, the \(I(\tilde{\nu })\) and \(R(\tilde{\nu })\) spectra are quite different and allowed the observation of an almost flat baseline [33, 34].
3 Results
To complete the characterization of the RE(III) ions for the whole lanthanide series (except promethium), it is necessary to consider dilute aqueous perchlorate solutions of europium, gadolinium, terbium and dysprosium. The peak positions of the entire series of the modes \(\nu_{1}\) Ln–(OH)2 for the rare earth ions, Ln3+(aq) including the ones in the middle of the series and those measured in previous studies [28,29,30] are given in Table 1. In addition to the \(\nu_{1}\) Ln–OH2 values, the Ln–O bond distances were also included in Table 1 for all the RE ions. In Figs. 1 and 2, the representative Raman spectra of dilute Eu(ClO4)3 and Gd(ClO4)3 solutions are presented. The \({\text{ClO}}_{4}^{ - } ({\text{aq}})\) modes have been given in recent publications (for details see refs [28, 34].).

Raman scattering profiles (Rpol (upper spectrum), Rdepol (middle spectrum) and Riso (lower spectrum) of a 0.370 mol·L–1 Eu(ClO4)3 solution. The depolarized perchlorate bands at 461 cm–1 and 628 cm–1 are the most prominent bands in the spectrum while the band of Eu–OH2 is broad and weak and only observed in the polarized scattering and is therefore present in the isotropic scattering. The inset gives the isotropic scattering profile in the terahertz region in more detail. The isotropic band at 367 cm–1 is assigned to the Eu–OH2 symmetric stretching mode

Raman scattering profiles Rpol (upper spectrum), Rdepol (middle spectrum) and Riso (lower spectrum) of a 0.403 mol·L–1 Gd(ClO4)3 solution. The depolarized perchlorate bands at 461 cm–1 and 628 cm–1 are prominent bands in the lower frequency part of the spectrum, while the band of Gd–OH2 is broad and weak and strongly polarized (ρ = 0.004) and therefore present in the isotropic scattering. The strongest \({\text{ClO}}_{4}^{ - }\)(aq) band at 933 cm–1, strongly polarized (ρ = 0.004), is due to the symmetric Cl–O stretch, ν1\({\text{ClO}}_{4}^{ - }\). The antisymmetric stretch of \({\text{ClO}}_{4}^{ - }\)(aq) is depolarized and appears as a broad band at 1112 cm–1. The inset gives the isotropic scattering profile in the terahertz region in more detail. The isotropic band at 374 cm–1 is assigned to the Gd–OH2 symmetric stretching mode
It is important to note that the \(\nu_{1}\) Ln–OH2 mode is also called the symmetric breathing modeFootnote 1since the oxygen atoms of the first hydration shell vibrate along the Ln–O bonds, stretching them in-phase. The molecule ion retains its symmetry and, therefore, the resulting dipole moment is zero. This normal mode depends on only one geometrical parameter: the radius r, the Ln–(OH2) bond distance. The breathing mode \(\nu_{1}\) Ln–OH2 of the rare earth ion hydrates was found to be almost completely polarized (ρ ~ 0) in solution, hence isotropic in character.
The peak positions of the totally symmetric stretching mode, ν1 Ln–(OH)2, for the rare earth ions in the middle of the series show a slight concentration effect which has been observed also for the light and heavy rare earth ions even though perchlorate is known as a weak complex forming anion [28,29,30, 35]. The concentration dependence of the \(\nu_{1}\) Ln–(OH2) mode of the RE ions in the middle of the series may be illustrated by the spectra of Gd(ClO4)3(aq) and Dy(ClO4)3(aq). This phenomenon was described for a variety of lanthanide solutions in our previous contributions [28,29,30, 35]. These earlier Raman spectroscopic contributions characterizing rare earth ions in aqueous perchlorate solutions showed that the solute concentration must be sufficiently small (~ 0.3 mol·L–1, which translates to a solute to water ration at ca. 1:182) in order to avoid ion pair formation. The concentration effect expresses itself in concentrated solutions as a slight downshift of the peak position on the \(\nu_{1}\) Ln–(OH2) mode and the subsequent broadening of its band width (full width at half height, fwhh). In concentrated solutions of Gd(ClO4)3(aq) larger than 2 mol·L–1, for instance, evidence for ion pair formation is found, although perchlorate is a weak complex forming anion. In these concentrated solutions there is simply not enough water to form complete hydration spheres of all ions (see [41] and discussions therein). The Raman spectrum of a 2.418 mol·L–1 Gd(ClO4)3 solution given in Fig. S1 shows the isotropic \(\nu_{1}\) Gd–OH2 mode which is slightly shifted to a lower peak position at 371 cm–1 compared with the one in a dilute solution where it appears at 374 cm–1 (Table 1). For the Dy(ClO4)3(aq) solution at 2.554 mol·L–1 (Fig. S2), a similar situation was observed, namely a shift of the ν1 Dy–(OH2) mode to slightly lower wavenumbers with a peak position at 381 cm–1 compared with the one for a 0.364 mol·L–1 Dy(ClO4)3(aq) with a \(\nu_{1}\) Dy–(OH2) mode at 384.5 cm–1 (Table 1). These peak shifts are also accompanied with a broadening of the band by ca. 15 cm–1. Furthermore, for an undisturbed observation of the \(\nu_{1}\) Ln–(OH2) mode, the nature of the counter ion is of importance. It may either act as weak complex forming anions such as perchlorate or trifluoromethansulfonate (triflate) or the ones with the tendency to form complexes such as chloride, nitrate and sulfate. In aqueous La2(SO4)3 solutions, which are quite dilute because of the sparingly soluble solute, the sulfato complex formation could, nevertheless, be detected in solutions as dilute as 10–3 mol·L–1. The breathing mode of the La3+ of the sulfato-complex, [La(OH2)nSO4]+, is down shifted by 31 cm–1 compared with the breathing mode of the non-complexed, fully hydrated [La(OH2)9]3+ species with a \(\nu_{1}\) value at 343 cm–1 [42].
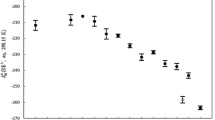
When following the breathing modes \(\nu_{1}\) Ln–OH2 for the RE ions of the whole series as a function of the atomic number z, an S-shaped curve results (Fig. S3). However, when giving the \(\nu_{1}\) Ln–OH2 breathing mode dependence as a function of their corresponding Ln–O bond distances an inverse shaped function results. This is simply due to the fact that with increasing atomic number of the RE ions the Ln–O bond distances of the RE ion hydrates decrease. The \(\nu_{1}\) Ln–OH2 breathing modes for the RE ions of the whole series as a function of the Ln–O bond distance, r is given in Fig. 3 and the RE ions are given on top of the abscissa of the graph. The rare earth ions Ln3+(aq) La3+, Ce3+, Pr3+ and Nd3+ form nona-hydrates and show the Ln–OH2 breathing modes at 343 cm–1, 344 cm–1, 347 cm–1 and 351 cm–1, respectively [30]. However, the \(\nu_{1}\) breathing mode for Sm3+(aq) appears at higher wavenumbers, namely at 362 cm–1 and it is thought to have a coordination number smaller than 9 namely 8.54 [15]. In addition, the Ln–OH2 breathing modes of the heavy rare earth ions Ho3+, Er3+, Tm3+, Yb3+ and Lu3+ were also characterized by Raman spectroscopy and these ions are known to form octa-hydrates [15,16,17] and their Ln–(OH2) breathing modes appeared at 387 cm–1, 389 cm–1, 391 cm–1, 394 cm–1, and 396 cm–1, respectively [31]. It is of interest that Y3+(aq), having properties similar to the heavy rare earth ions dysprosium and holmium, is coordinated by eight water molecules in the first coordination sphere forming a square antiprismatic coordination polyhedron. The \(\nu_{1}\) Y–(OH2) breathing mode for [Y(OH2)8]3+ appears at 384 cm−1 [36] and with the ionic radius of Y3+(aq) at 2.36 Å [36] plots close to the one for \(\nu_{1}\) Dy–OH2 (see Fig. 3).

[37]
The totally symmetric stretching modes, ν1 Ln–OH2, of the trivalent lanthanide ions, [Ln(OH2)n]3+ with n = 9, 8 and 9/8 (Ln = La–Lu, except Pm) as a function of the Ln–OH2 bond distance of their corresponding hydrates. The solid curve represents the fitted curve {a + b·tanh(c(r – rm))} of ν1 Ln–OH2 data (R2 = 0.984) as a function of r, the Ln–OH2 bond distances of the hydrates. The open square shows the ν1 Y–OH2 peak position of the octahydrate, [Y(OH2)8]3+(aq) [36]. The Ln–OH2 bond distances including the one for Y-OH2 were taken from ref
The individual band parameters for all the \(\nu_{1}\) Ln–(OH2) modes of the Ln3+(aq) ions are given in Table 1, specifically the scattering intensity of these bands, fwhh and the force constants of the modes. Additionally, the Ln–OH2 bond distances (r) and kinetic parameters regarding the H2O exchange of the water molecules in the first sphere are presented. Our spectroscopic results will now be discussed in detail together with these parameters on Ln3+(aq) ions. High level quantum mechanical calculations in recent works and our own calculations will be taken into account to shed light upon the structure of the rare earth ion hydrates in solution.
4 Discussion of the Raman Spectroscopic Results
The peak position of the \(\nu_{1}\) Ln–(OH2) modes of the rare earth ions given as a function of the Ln–O bond distances, r,Footnote 2) (Table 1) may be expressed by Eq. 4:
and the following parameters were obtained: a = 380.5 cm–1, b = 32.35 cm–1, c = –8.97 Å–1 and rm = 2.435 Å. As previously mentioned, an inverse S-shaped dependence appears as a function of their Ln–OH2 bond distances given in Fig. 3. The \(\nu_{1}\) Ln–(OH2) breathing modes show a drastic change between samarium and terbium. However, then the \(\nu_{1}\) Ln–(OH2) frequencies depend upon the atomic number z of the RE(III) ions following an S-shaped curve (Fig. S3). The light rare earth ions forming nona-hydrates in aqueous solution plot on an almost straight line beneath the one for the heavy rare earth octa-hydrates. The difference in wavenumbers between LRE ions and HRE ions is ca. 18 cm–1 and the ions in the middle of the series are displayed between these two lines. The first derivative of the Eq. 4 describing the relationship of \(\nu_{1}\) Ln–(OH2) mode as a function of Ln–O bond distance, r, was taken and shows how much an error in bond distance with ± 0.02 Å affects the frequencies of the modes of the rare earth ion hydrates, \(\nu_{1}\) Ln–(OH2). The derivative of Eq. 4 leads to Eq. 5:
and the relation \({\Delta }\nu_{{1}} {(}r{) } \approx \, \frac{{{\text{d}}\nu_{{1}} {(}r)}}{{{\text{d}}r}} \cdot {\Delta }r\) allows one to estimate the uncertainty \({\Delta }\nu_{{1}} {(}r{)}\) and to determine the frequency of the Ln–O mode when the uncertainty \({\Delta }r\) in determining the Ln–O bond distances is given. For the light and heavy rare earth ions, an uncertainty of ± ~ 3 cm–1 follows and for the intermediate ions ± 6 cm–1. The uncertainties in determining the peak positions of the \(\nu_{1}\) Ln–(OH2) of the rare earth ions are ± 1 cm–1 and therefore are much smaller than the above calculated uncertainties. In other words, the symmetric breathing modes reflect the change in the first coordination numbers across the lanthanide series much more sensitively than methods determining the Ln–(OH2) bond distances. The reason for that is the Raman frequencies depend not only on the bond distance but also on the reduced mass of the ligand.
The scattering intensity (relative integrated molar scattering intensity, Sh) of the \(\nu_{1}\) Ln–(OH2) modes is very small and with their broad band profiles makes them difficult to detect by Raman spectroscopy (Table 1). The relative integrated molar scattering intensity, Sh, is defined by Eq. 6 as:
where \(A_{{{\text{Ln}}^{{3 + }} }}\) and \(A_{{{\text{ClO}}_{{4}}^{ - } }}\) are integrated band intensities from R- spectra for the \(\nu_{1}\) Ln–OH2 bands and the \(\nu_{1}\)\({\text{ClO}}_{4}^{ - }\) band respectively. \(C_{{{\text{Ln}}^{{3 + }} }}\) and \(C_{{{\text{ClO}}_{{4}}^{ - } }}\) are the molar concentrations of the Ln3+ ions and the perchlorate ions and Cinstr is a constant which corrects for the instrumental factors. The Sh values for the lanthanide ion hydrates are small compared to softer more polarizable ions such as In3+ [43]. It is known that the lanthanide ions are hard cations and therefore less polarizable ions [44].
The bandwidths of the isotropic \(\nu_{1}\) Ln–OH2 bands are quite broad and are inhomogeneously broadened which is known as environmental broadening. In solution, the local environment varies significantly and slightly different spectrum will result and so the overall spectral shape is significantly broadened. Molecular dynamics (MD) results [45, 46] demonstrated the flexibility of the lanthanide hydrate structures such as minor ultrafast geometric modifications within the hydrate or from pseudorotations, requiring substantially longer times in the scale at ca. 1 ps for establishment of an altered geometry [45]. The vibrational periods of the \(\nu_{1}\) Ln–OH2 vibrations range from 0.097 ps for La3+ to 0.082 ps for Lu3+ [30, 31, 35] so that the modes vibrate several hundred thousand times before the hydrated Ln3+ species experiences a water exchange. (The establishment of slightly altered geometry according to Rode et al. [45, 46] is of the order of a ps and allows only a few vibrational periods to occur.)
Water exchange reactions for the [Ln(OH2)n]3+ species in aqueous solution (see Table 1) were reported from ultrasound absorption measurements [38] and more recently from NMR studies [39, 40] (Table 1). From water exchange rates \(k_{i}^{298}\) of all the Ln3+(aq) ions (interchange rate of an inner-sphere water molecule by a \({\text{SO}}_{4}^{2 - }\) ion [38]), it follows that the exchange rates \(k_{i}^{298}\) start at smaller values with La3+ at 2.1 × 108 s–1, reaching a maximum at Sm3+ (7.4 × 108 s–1), remaining at high values in going to Tb3+ (ions in the middle of the series) and then falling rapidly reaching Lu3+ with a value at 6 × 107 s–1. (More recent data on the rates of water exchange, \(k_{{{\text{ex}}}}^{298}\) measured directly by NMR [39, 40] correlate closely with the \(k_{i}^{298}\) values for the RE ions reassuring the validity of the data in [38]). It is worthwhile to mention that recently a plethora of MD results (see for instance [45, 46] and references therein) have been published giving structural data for the hydrated rare earth ions and also dynamic parameters. However, the water exchange rates put forward by these theoretical simulations are very short; actually too short to justify the aforementioned experimental values. It is clear that the water exchange kinetic in aqueous solution between first sphere water and second sphere water is a diffusion controlled process and therefore of limited rate (see Eigen [47]).
The force constant calculations for the \(\nu_{1}\) Ln–OH2 breathing modes of the Ln(III) hydrate species (Table 1), applying a simple model, taking only one ligand into account, have been carried out according to Eq. 7:
with c, the velocity of light, \(\tilde{\nu }_{i}\) the wavenumber of the mode i, N the Avogadro constant and AL the molecular weight of the ligand, in our case, water. Such a simplification is justifiable because the totally symmetric character of the \(\nu_{1}\) Ln–OH2 mode dictates that the central cation, Ln3+ remains stationary and only the water molecules are involved in the breathing motion along the bonds without disturbing the symmetry and therefore the mode is totally polarized. The force constants, kLn–O, calculated for the measured \(\nu_{1}\) Ln–OH2 breathing modes, and the corresponding Ln3+–O bond distances [37] are given in Table 1. The force constants increase in going from La3+ to Lu3+ form an S-shaped curve plotted against the atomic number of the rare earth ion which is shown in Fig. S3. The ions of the light rare earths (LRE) plot on as almost linear curve as do the heavy rare earth (HRE) ions which are displayed also on a linear curve offset to the former and with the ions in the middle of the series (Sm–Tb) appearing between the two lines (see Fig. 4). The LRE ions forming nona-hydrates, the heavy ones octa-hydrates and the RE ions in the middle show an overall hydration number between 9 and 8, with non integer coordination numbers. It should be mentioned that this S-shape behavior has been observed for a variety of thermodynamic properties of these ions and has been published for quite some time [7,8,9]. The assumed break of the properties between the light and heavy rare earths does not mean that the hydration number drops suddenly in the middle of the series but a dynamic hydration change from 9 to 8 may be envisioned. Such a dynamic structure has been presented recently by a quantum chemical (QC) study [22] taking into account two aspects, energy and wave function.

Top panel: Peak positions of the Ln–OH2 breathing modes plotted against the atomic number of the rare earth ions across the whole series. Furthermore, the DFT frequencies for the nonahydrate of La3+ and its (hypothetical) octa-hydrate are given as well as for the octa-hydrate of Lu3+ and its (hypothetical) nona-hydrate. The black line represents the frequencies for the octa-hydrates and the broken line the ones for the nona-hydrates. Bottom panel: Experimental Ln–OH2 bond distances of the [Ln(OH2)n]3+ (n = 9, 8 and 9/8) hydrates. The open squares show the data from ref [37]. and the black filled squares refs [15,16,17]. from the ND first order technique
The QC analysis by Zhang et al. [22] contributed greatly to the understanding of the structure and kinetics of the Ln3+(aq) ions across the whole lanthanide series. From their wave function analysis including the atoms in molecules approach by Bader (see ref. in [22]) it was shown that the nature of the Ln3+–oxygen bonds are electrostatic with very small covalent contributions. A specific behavior was found regarding the capping Ln−O bonds for the RE ions across the whole lanthanide series. The bond strength for the tri-capped water molecules is strong at the beginning of the series for the nona-hydrates form lanthanum to samarium. But going from samarium to lutetium, the capping Ln−O bonds in the nona-aqua lanthanide hydrates become weaker and, unexpectedly, shorten. Finally, as the capping Ln−O bonds are getting easier to disrupt, heavier lanthanides will prefer a CN between 9 and 8.Footnote 3 This model suggests that the capping Ln−O bonds of moderate strength, which occur for intermediate lanthanides, are advantageous for the formation of an intermediate bi-capped trigonal prism (BTP) during water exchange and explains the kinetic of the water exchange rate reaching a maximum for the intermediate region of the rare earth ions but is lowest at the beginning and end of the series. This behavior of the capping Ln−O bonds explains not only the kinetics of the Ln3+ hydrates across the series but also the structures and energetics of the Ln3+ hydrates [22]. As a result, the hydrates of the lanthanide ions across the whole series form stable nona-hydrates at the beginning of the series (lanthanum to promethium), the heavy ones form octa-hydrates while the intermediate rare earth ions have a non integer coordination number between 9 and 8 according to the ratio of nona- and octa-aqua hydrates. Such a peculiar behavior of capping Ln−O bonds put forward by Zhang and Dolg [22] determining the hydration behavior of Ln3+ ions across the whole series is also portrayed by the course of the totally symmetric stretching mode, \(\nu_{1}\) Ln–OH2 and their respective force constants across the series (Table 1 and Fig. S3) and reinforces these quantum mechanical model findings.
In the following, we will compare our experimental results on the \(\nu_{1}\) Ln–OH2 breathing modes across the series (Figs. 4, 5) with results from DFT calculations. However, the large number of electrons, partly filled 4f shells, and relativistic effects of Ln3+ ions make quantum mechanical calculations difficult [21,22,23, 49]. It should be pointed out that Zhang et al. [22] used different methods such as B3LYP, (SCS)-MP2, and CCSD(T) for the water cluster calculations in the inner shell and COSMO model for simulating the outer water shells. Reliable first principle values on hydration Gibbs energies were reported which correspond well with the experimental data and the corresponding coordination numbers 9, 8 and intermediate ones for light, heavy and intermediate rare earth ions. However, the geometry optimizations were restricted to gas phase clusters where the MP2 method was used but frequencies of the normal vibrations of the clusters were, unfortunately, not reported. The Ln–OH2 bond distances of naked clusters are systematically overestimated in comparison with the experimental ones due to the neglect of the surrounding water molecules beyond the first hydration shell. As a result, vibrational frequencies are, therefore, underestimated. Due to many saddle points on the potential energy surface and convergence problems [49] it proved difficult to perform Ln–OH2 cluster geometry optimizations within continuum models while taking higher hydration shells into account.

Given are the calculated bond distances for the nona- and octa-hydrates using the corresponding effective ionic radii for coordination 9 and 8, respectively [51], and adding the radius for the water molecules [52]. Upper (broken) line and red small data points correspond to the data of the nona-hydrates and below the small blue data points and the black line correspond to the octa-hydrates. The experimental Ln–OH2 bond distances of the [Ln(OH2)n]3+ (n = 9 or 8) hydrates are also given: filled triangle EXAFS [37], filled square XRD [10,11,12], inverted triangle ND [13] + ND [14], x ND [16]. Furthermore, the DFT Ln–OH2 bond distances (Table 2) are given for the nona-hydrate of La3+ and its (hypothetical) octa-hydrate as well as for the octa-hydrate of Lu3+ and its (hypothetical) nona-hydrate (Color figure online)
We obtained stable geometries without imaginary frequencies for the clusters {[La(H2O)9]3+}PCM and {[Lu(H2O)8]3+} PCM by applying the polarized continuum model (PCM) [50] with self-consistent field (SCF) procedure and starting without geometrical constraints. The presence of the tricapped trigonal structure (TTS) and square antiprism structure (SAP) at the beginning and the end of the Ln3+ series, respectively, was established and this is generally accepted. The B3LYP functional and Stuttgart/Dresden basis set SDD with effective core potential were used [51]. In Table 2, the Ln–OH2 bond distances of the clusters {[La(H2O)9]3+} PCM and {[Lu(H2O)8]3+}PCM agree well (scaling factors 1.006/1.000) with the experimental values, as do the frequencies of the breathing vibration \(\nu_{1}\) (scaling factors 0.989/0.983). Control calculations with the Los Alamos basis set LANL2DZ for the La3+-nona cluster gave a value at 328.2 cm−1 and a mean bond distance at 2.565 Å. As pointed out, the TTP structure has six equidistant bonds to the water oxygen atoms in the prism (p) and three to the capping (c) position, and the mean bond distance m (m) denotes their mean value. Applying PCM, however, to the (hypothetical) clusters [La(H2O)8]3+ or [Lu(H2O)9]3+ also lead to unsuccessful results (compare results in [21]). Therefore, we used the scaling factors from the gas phase clusters of [La(H2O)9]3+ and [Lu(H2O)8]3+ (Table 2). It should be pointed out that all normal vibrations of the naked clusters were without imaginary frequencies. With respect to the Ln–O bond distances and the breathing frequencies, two effects must be taken into account which we estimated and introduced in Table 2 and Fig. 4. Beginning with cerium and the elements that follow with increasing atomic number z, the 4f shell is successively filled with electrons shielding the central positive charge and diminishing the lanthanide ion radius by about 0.02 Å between neighboring ions for both cluster series, namely the nona-coordinated hydrates and the octa-coordinated ones (Fig. 5). This effect is known as the lanthanide contraction. Taking Shannon’s effective radii for the coordination numbers 9 and 8 [52] and adding the mean water molecule radius at 1.34 Å [53] results in the Ln–OH2 bond distances for the whole series (Fig. 5). The experimental bond distances appear at or between these two lines with the light rare earth ion hydrates at the line for the nona-hydrates, the heavy rare earths on the one for octa-hydrates and the intermediate ones between the two lines taking the considerable uncertainty of the bond distances into account. Secondly, a change from coordination number 9 to CN 8 results in a reduced Ln3+–O bond distance by ca. 0.04 Å for La3+ and by 0.05 Å for Lu3+ (Table 2). Decreasing the Ln–O bond distance, however, is accompanied by a frequency increase of the breathing vibration, \(\nu_{1}\) Ln–(OH2). Changing the CN from 9 to 8 results in a frequency increase of \(\nu_{1}\) by ca. 14 cm–1 in going from [La(OH2)9]3+ to the (hypothetical) [La(OH2)8]3+ and 20 cm–1 frequency decrease in going from [Lu(OH2)8]3+ to the (hypothetical) [Lu(OH2)9]3+. Along the two straight lines in Fig. 5 for the nona-hydrates and the octa-hydrates, this corresponds to a ca. 2.4 cm–1 frequency difference \(\Delta \nu\) for the LRE and a ca. 2.8 cm–1\(\Delta \nu\) for the HRE between neighboring ions. It should be pointed out that this frequency difference between nona- and octa-hydrates is much larger than the uncertainty in determining \(\nu_{1}\) with an accuracy of ± 1 cm–1 [30, 31, 35] and, therefore, reflects the coordination change well.
5 Conclusion
The isotropic Raman bands of the \(\nu_{1}\) Ln–OH2 modes of the rare earth ions in aqueous solution, [Ln(OH2)n]3+ (n = 9,8 and 9/8) as a function of the Ln–OH2 bond distance across the whole series show an S-shaped behavior. The light rare earth ions Ln3+(aq) (Ln = La3+ to Nd3+) plot on a straight line as do the heavy rare earth ions (Dy3+, Ho3+, Er3+, Tm3+, Yb3+ and Lu3+) which plot on a straight line above the one for the LRE ions. The middle rare earth ions (Sm3+, Eu3+, Gd3+ and Tb3+) plot inbetween. For the LRE ions which are known to be nona-hydrates, the oxygen atoms form a tricapped trigonal prismatic structure and the isotropic bands of these hydrates increase in an almost linear fashion with increasing atomic number, whilst the octa-hydrates of the HRE ions plot on a much higher straight line and the wavenumber difference between the nona- and octa-hydrates is ca. 18 cm–1. Again, the \(\nu_{1}\) Ln–O frequencies of the Raman bands of the MRE ions plot in between these two straight lines. The S-shaped behavior found for the \(\nu_{1}\) Ln–OH2 modes plotted against the atomic number for the hydrates of the rare earth ions across the whole series is in agreement with data on apparent molal volumes, relative viscosities, and molal heat capacities. No abrupt gadolinium break could be observed but a change of the coordination number from 9 to 8 in the middle of the series was detected. Recent QC results [22] explain the Raman data across the lanthanide series in a qualitative way but contradict the view of the specific asymmetry of the capping waters in the TTP structure of the nona-hydrate structure across the lanthanide series with water deficient configurations for the smallest ions (see [18] p. 3061–3062).
Notes
The published Ln-OH2 bond distances vary several tenths of an Å which is outside the expected accuracy at ± 0.02 Ẳ [17]. Measurements via the ND first order technique [15,16,17] may serve as benchmark. The nature of the counter ions may be a problem especially in concentrated solutions because the frequently used anions such as Cl–, \({\text{NO}}_{3}^{ - }\) and \({\text{SO}}_{4}^{2 - }\) form complexes with RE ions. Recent Raman spectroscopic measurements on a variety of RE ions in \({\text{ClO}}_{4}^{ - }\) solutions confirmed this point [28,29,30]. Furthermore, method specific problems may also contribute to the uncertainty of the Ln–OH2 bond distances.
The QC analysis in [22] showed convincingly that the postulated asymmetry in bond strength for the capping bonds (ref.[48]) for the heavier lanthanides starting from samarium cannot be verified and no single capping bond plays a special role. Therefore the model put forward in ref.[48] may be questionable although it is undoubtable true that no abrupt structural change occurs at gadolinium (ref.[48], p. 7706).
References
Huang, C.-H., Bian, Z.: Introduction. In: Huang, C.-H. (ed.) Introduction in Rare Earth Coordination Chemistry: Fundamentals and Applications, pp. 1–39. Wiley, Singapore (2010)
Goldschmidt, V.M., Barth, T., Lunde, G.: Geochemische Verteilungsgesetze der Elemente. 5. Isomorphie und Polymorphie der Sesquioxyde: Die Lanthaniden-Kontraktion und ihre Konsequenzen. Kristiania Ja, Dybwad (1925)
Spedding, F.H., Pikal, M.J., Ayers, B.O.: Apparent molal volumes of some aqueous rare earth chloride and nitrate solutions at 25°. J. Phys. Chem. 70, 2440–2449 (1966)
Spedding, F.H., Jones, K.C.: Heat capacities of aqueous rare earth chloride solutions at 25°. J. Phys. Chem. 70, 2450–2455 (1966)
Spedding, F.H., Pikal, M.J.: Relative viscosities of some aqueous rare earth chloride solutions at 25°. J. Phys. Chem. 70, 2430–2440 (1966)
Spedding, F.H., Rard, J.A., Saeger, V.W.: Electrical conductances of some aqueous rare earth electrolyte solutions at 25 °C. II. Rare earth chlorides. J. Chem. Eng. Data. 19, 373–378 (1974)
Rizkalla, E.N., Choppin, G.R.: Hydration and hydrolysis of lanthanides. In: Gschneidner Jr., K.A., Eyring, L. (eds.) Handbook on the Physics and Chemistry of Rare Earths, pp. 393–442. Elsevier, Amsterdam (1991)
Fourest, B., Duplessis, J., David, F.: Comparison of diffusion coefficients and hydrated radii for some trivalent lanthanide and actinide ions in aqueous solution. Radiochim. Acta 36, 191–195 (1984)
David, F., Vokhmin, V., Ionova, G.: Water characteristics depend on the ionic environment. Thermodynamics and modelisation of the aquo ions. J. Mol. Liq. 90, 45–62 (2001)
Habenschuss, A., Spedding, F.H.: The Coordination (hydration) of rare earth ions in aqueous chloride solutions from X ray diffraction. I. TbCl3, DyCl3, ErCl3, TmCl3, and LuCl3. J. Chem. Phys. 70, 3758–3763 (1979)
Habenschuss, A., Spedding, F.H.: The coordination (hydration) of rare earth ions in aqueous chloride solutions from X ray diffraction. II. LaCl3, PrCl3 and NdCl3. J. Chem. Phys. 70, 2797–2806 (1979)
Habenschuss, A., Spedding, F.H.: The coordination (hydration) of rare earth ions in aqueous chloride solutions from X ray diffraction. III. SmCl3, EuCl3, and series behavior. J. Chem. Phys. 73, 442–450 (1980)
Narten, A.H., Hahn, R.L.: Hydration of the Nd3+ ion in neodymium chloride solutions determined by neutron diffraction. J. Phys. Chem. 87, 3193–3197 (1983)
Annis, B.K., Hahn, R.L., Narten, A.H.: Hydration of the Dy3+ ion in dysprosium chloride solutions determined by neutron diffraction. J. Chem. Phys. 82, 2086–2091 (1985)
Cossy, C., Helm, L., Powell, D.H., Merbach, A.E.: A change in coordination number from nine to eight along the lanthanide(lll) aqua ion series in solution: a neutron diffraction study. New J. Chem. 19, 27–36 (1995)
Helm, L., Foglia, F., Kowall, T., Merbach, A.E.: Structure and dynamics of lanthanide ions and lanthanide complexes in solution. J. Phys. 6, A132–A140 (1994)
Cossy, C., Barnes, A.C., Enderby, J., Merbach, A.E.: The hydration of Dy3+ and Yb3+ in aqueous solution: a neutron scattering first order difference study. J. Chem. Phys. 90, 3254–3260 (1989)
Persson, I., D’Angelo, P., De Panfilis, S., Sandström, M., Eriksson, L.: Hydration of lanthanoid(III) ions in aqueous solution and crystalline hydrates studied by EXAFS spectroscopy and crystallography: the myth of the “gadolinium break”. Chem. Eur. J. 14, 3056–3066 (2008)
D'Angelo, P., Spezia, R.: Hydration of lanthanoids(III) and actinoids(III): an experimental /theoretical saga. Chem. Eur. J. 18, 11162–11178 (2012)
Spezia, R., Duvail, M., Vitorge, P., D’Angelo, P.: Molecular dynamics to rationalize EXAFS experiments: a dynamical model explaining hydration behaviour across the lanthanoid(III) series. J. Phys: Conf. Series 190, 1–6 (2009). https://doi.org/10.1088/1742-6596/190/1/012056
Clark, A.E.: Density functional and basis set dependence of hydrated Ln(III) properties. J. Chem. Theory Comput. 4, 708–718 (2008)
Zhang, J., Heinz, N., Dolg, M.: Understanding lanthanoid(III) hydration structure and kinetics by insights from energies and wave functions. Inorg. Chem. 53, 7700–7708 (2014)
Ciupka, J., Cao-Dolg, X., Wiebke, J., Dolg, M.: Computational study of lanthanide(III) hydration. Phys. Chem. Chem. Phys. 12, 13215–13223 (2010)
Peppard, D.F., Mason, G.W., Lewey, S.: A tetrad effect in the liquid–liquid extraction ordering of lanthanide(III). J. Inorg. Nucl. Chem. 31, 2271–2272 (1969)
Laing, M.: Gadolinium: central metal of the lanthanoids. J. Chem. Educ. 86, 188–189 (2009)
Nugent, L.J.: Theory of the tetrad effect in the lanthanide(III) and actinide(III) series. J. Inorg. Nucl. Chem. 32, 3485–3490 (1970)
Müller, D., Knoll, C., Herrmann, A., Savasci, G., Welch, J.M., Artner, W., Ofner, J., Lendl, B., Giester, G., Weinberger, P., Steinhauser, G.: Azobis[tetrazolide]-carbonates of the lanthanides—breaking the gadolinium break. Eur. J. Inorg. Chem. 2018, 1969–1975 (2018). https://doi.org/10.1002/ejic.201800218
Rudolph, W.W., Irmer, G.: Hydration and ion pair formation in common aqueous La(III) salt solutions—a Raman scattering and DFT study. Dalton Trans. 44, 295–305 (2015). https://doi.org/10.1039/c4dt03003f
Rudolph, W.W., Irmer, G.: Raman spectroscopic characterization of light rare earth ions: La3+, Ce3+, Pr3+, Nd3+ and Sm3+—hydration and ion pair formation. Dalton Trans. 46, 4235–4244 (2017)
Rudolph, W.W., Irmer, G.: On the hydration of heavy rare earth ions: Ho3+, Er3+, Tm3+, Yb3+ and Lu3+—a Raman study. Molecules 24(1953), 1–18 (2019). https://doi.org/10.3390/molecules24101953
Schwedt, G., Schmidt, T.C., Schmitz, O.J.: Analytische Chemie: Grundlagen Methoden und Praxis. Dritte Überarbeitete Auflage. Wiley, Weinheim (2016)
Rudolph, W.W., Irmer, G.: Raman and infrared spectroscopic investigations on aqueous alkali metal phosphate solutions and density functional theory calculations of phosphate–water clusters. Appl. Spectrosc. 61, 1312–1327 (2007). https://doi.org/10.1366/000370207783292037
Rudolph, W.W., Irmer, G.: Vibrational spectroscopic studies and density functional theory calculations of speciation in the CO2–water system. Appl. Spectrosc. 60, 130–144 (2006)
Rudolph, W.W., Irmer, G.: Hydration of the Ca(II) in an aqueous solution of common anions (, Cl−, Br−, and ). Dalton Trans. 42, 3919–3935 (2013)
Rudolph, W.W., Irmer, G.: Hydration and ion pair formation in aqueous Lu3-solution. Molecules 23, 3237 (2018). https://doi.org/10.3390/molecules23123237
Rudolph, W.W., Irmer, G.: Hydration and ion pair formation in aqueous Y3+–salt solutions. Dalton Trans. 44, 18492–18505 (2015). https://doi.org/10.1039/c5dt02586a
Persson, I.: Hydrated metal ions in aqueous solution: how regular are their structures? Pure Appl. Chem. 82, 1901–1917 (2010)
Fay, D.P., Litchinsky, D., Purdie, N.: Ultrasonic absorption in aqueous salts of the lanthanides. J. Phys. Chem. 73, 544–552 (1969)
Helm, L., Nicolle, G.M., Merbach, A.E.: Water and proton exchange processes on metal ions. Adv. Inorg. Chem. 57, 327–379 (2005)
Helm, L., Merbach, A.E.: The periodic table and kinetics? Chimia 73, 179–184 (2019)
Marcus, Y.: On water structure in concentrated salt solutions. J. Solution Chem. 38, 513–516 (2009)
Rudolph, W.W., Irmer, G.: A Raman spectroscopic investigation of speciation in La2(SO4)3(aq). RSC Adv. 5, 84999 (2015). https://doi.org/10.1039/C5RA16900C
Rudolph, W.W., Fischer, D., Tomney, M.R., Pye, C.C.: Indium(III) hydration in aqueous solutions of perchlorate, nitrate and sulfate. Raman and infrared spectroscopic studies and ab-initio molecular orbital calculations of indium(III)–water clusters. Phys. Chem. Chem. Phys. 6, 5145–5155 (2004)
Pearson, R.G.: Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539 (1963)
Lutz, O.M.D., Hofer, T.S., Randolf, B.R., Rode, B.M.: Computational study of the cerium(III) ion in aqueous environment. Chem. Phys. Lett. 539–540, 50–53 (2012)
Tirler, O., Passler, P.P., Rode, B.M.: The lanthanoid hydration properties beyond the ‘gadolinium break’: dysprosium(III) and holmium(III), an ab initio quantum mechanical molecular dynamics study. Chem. Phys. Lett. 635, 120–126 (2015)
Eigen, M.: Fast elementary steps in chemical reaction mechanisms. Pure Appl. Chem. 6, 97–115 (1964)
Abbasi, A., Lindqvist-Reis, P., Eriksson, L., Sandstroem, D., Lidin, S., Persson, I., Sandstroem, M.: Highly hydrated cations: deficiency, mobility, and coordination of water in crystalline nonahydrated scandium(III), yttrium(III), and lanthanoid(III) trifluoromethanesulfonates. Chem. Eur. J. 11, 4065–4077 (2005)
Kuta, J., Clark, A.E.: Trends in aqueous hydration across the 4f period assessed by reliable computational methods. Inorg. Chem. 49, 7808–7817 (2010). https://doi.org/10.1021/ic100623y
Cossi, M., Scalmani, G., Rega, N., Barone, V.: New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 117, 43–54 (2002). https://doi.org/10.1063/1.1480445
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.A.J.T., Vreven, K.N., Kudin, J.C., Burant, J.M., et al.: Gaussian 03, Revision, C.02. Gaussian Inc., Wallingford (2004)
Shannon, R.D.: Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 32, 751–767 (1976)
Beattie, J.K., Best, S.P., Skelton, B.W., White, A.H.: Structural studies on the caesium alums, CsMIII[SO4]2·12H2O. J. Chem. Soc. Dalton Trans. (1981). https://doi.org/10.1039/DT9810002105
Acknowledgments
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rudolph, W.W., Irmer, G. On the Hydration of the Rare Earth Ions in Aqueous Solution. J Solution Chem 49, 316–331 (2020). https://doi.org/10.1007/s10953-020-00960-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-020-00960-w