
Abstract
In this work, a recycled bis(2-hydroxyethyl) terephthalate (BHET) monomer, obtained by glycolysis of marine polyethylene terephthalate (PET) litter, and a biobased polyol derived from castor oil were used for the synthesis of thermoset polyurethanes (PUs). BHET was obtained in a closed reactor at 220 °C and a short reaction time of 30 min. Different PUs were synthesized, varying the BHET content, ranging from 0 to 21 wt% and varying the polyol content, obtaining a renewable carbon content between 23 and 43%. The PUs synthesized in this work, in which at least 40% of their components are renewable and recycled, showed an interesting combination of thermal, thermo-mechanical and mechanical properties. In addition, a preliminary chemical recycling study of the synthesized PUs was performed to evaluate their recyclability, wrapping the whole process within the circular economy. The obtained glycolyzed product was a single-phase viscous liquid consisting on polyol-rich and BHET-rich fractions, with contents in the range of those employed in the synthesis of PUs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nowadays, the world is facing one of the biggest environmental problems related to the unstoppable production and massive use of plastic materials. The abundant annual global plastic production, estimated at 360 million tons [1], generates significant amounts of plastic waste that accumulate in landfills and in natural environments, including marine and terrestrial ecosystems [2]. Therefore, environmentally friendly materials, together with an adequate waste treatment system, are needed to move from a linear economy system to a circular economy one [3].
Currently, one of the main environmental problems is the dumping of non-biodegradable fossil plastics in marine ecosystems, leading to the formation of floating islands, as well as the production of plastic debris and fragments, such as microplastics, which affect surface waters and living animals [4]. As plastics usually have a high physical and chemical stability, their natural degradation can last for thousands of years in marine environment [5]. It is estimated that each year between 4.8 and 12.7 million tons of plastic end up in the sea [6], the most common plastics found in the sea are polyethylene (PE), polypropylene (PP) and polyethylene terephthalate (PET) [7, 8].
PET, with a production of 70 million tons per year, is one of the most produced plastics in the world, occupying the fifth place in the world production ranking [2, 9]. This polyester has interesting properties for the packaging sector, making it the main polymer used for packaging mineral water, beverages and oils [10]. Due to its widespread use, a large amount of PET waste is generated each year, proximally 4.6 million tons, and it is necessary to properly manage this residue [9].Thermo-mechanical recycling is nowadays the most widely used method for PET waste, due to its simplicity and low cost. This method was implemented during the year 1977 [11]. However, it is limited to the recycling of lightly degraded waste, such as post-industrial and urban waste, which is not the case for marine PET litter. In addition to being a major environmental problem and requiring a much more complicated collection process, the marine PET litter is also of poorer quality, as it undergoes more severe degradation in the sea [12]. For this reason, recycling techniques used for PET urban waste are not suitable for the treatment of marine litter. Chemical recycling of marine PET bottles through glycolysis was successfully developed in a previous work, obtaining high purity bis(2-hydroxyethyl) terephthalate (BHET) in a closed reactor at low reaction times [13]. BHET could be used again for the synthesis of PET [14] or as raw material for the production of different polymers [15], such as polyurethanes [16].
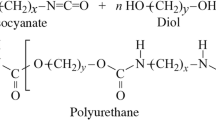
Polyurethanes (PU) are one of the most abundant plastics in the world, with a global production of 27 million tons per year [17], ranking seventh in world production [9]. Their versatility allows them to be used in a wide range of applications such as in packaging, construction, textiles, thermal and acoustic insulation, and automotive and biomedical applications [18,19,20,21]. Moreover, as for PET, the chemical recyclability of PUs has also been demonstrated, from which monomers or building blocks suitable for synthesizing new polymers can be obtained [22,23,24,25].
In recent years, the synthesis of a wide variety of polyurethanes containing BHET recovered from municipal PET waste has been reported, for the obtaining of foams, adhesives, coatings and elastomers, among others [26,27,28,29,30,31]. In a recent work, we have developed polyurethane elastomers using BHET obtained from the recycling of marine PET litter as chain extender [32]. The materials obtained exhibit physicochemical, thermal and mechanical properties comparable to those synthesized from commercial fossil-derived BHET.
Therefore, this work aims the synthesis of recycled thermoset polyurethanes by incorporating the BHET monomer obtained from the glycolysis of marine PET litter. Moreover, in order to develop increasingly sustainable materials, a polyol derived from castor oil was incorporated into the polyurethane formulation. The synthesis of the new thermoset polyurethane targets more than 40% of its content to be renewable and/or recycled. Moreover, the chemical recycling of the synthetized PU was carried out, in order to prove the recyclability of PU. The objective of this study is threefold: first, the valorization of waste, incorporating recycled BHET from marine PET litter in the synthesis of PUs; second, to develop high performance materials, thermoset polyurethanes of high modulus and strength; finally, to demonstrate the recyclability of the synthesized thermosets.
Materials and Methods
Materials
The BHET used in this work was obtained from the glycolysis of marine PET litter, following the previously published protocol and named as BHET-m [12]. It is solid at room temperature (Tm = 110 ºC), it presents a hydroxyl index (IOH) of 452 mg KOH g−1, a functionality of 2, and molecular weight of 254 g mol−1. For the synthesis of thermoset polyurethanes (PU), a castor oil derived polyol with an 80% of renewable carbon content purchased from Vertellus (Denham Springs, USA) (Polycin 12, functionality = 4, IOH = 330 mg KOH g−1 and viscosity = 300 mPa s) was used. On the other hand, as isocyanate a commercial aromatic polymeric diphenyl methane diisocyanate (pMDI) supplied by Covestro (Germany) (Desmodur 44 V, NCO equivalent weight = 131.3 g/eq and viscosity = 160–240 mPa s) was used. Prior to use, the polyol was dried under vacuum at 65 °C for 4 h.
Synthesis of Thermoset Polyurethane
Polyurethanes with different Polyol:BHET-m:pMDI ratios were synthesized in an one-step polymerization process without catalysts. BHET-m is solid at room temperature and insoluble in the polyol, so it was first, mixed with pMDI in a 250 mL round-bottom flask equipped with a mechanical stirrer for about 15–20 min until a uniform mixture was observed. Then, the previously dried polyol was added to the mixture and it was stirred vigorously for 5–10 min until a homogeneous mixture was obtained. Subsequently, the reaction mixture was casted between two Teflon coated metal plates separated by 1.5 mm and pressed under 50 bar, first at 120 °C for 2 h, and then at 140 °C for 2 h. A fraction of the mixture was separated to analyze the curing reaction by differential scanning calorimetry (DSC). The isocyanate index, defined as the ratio between the isocyanate equivalents used in the formulation and the stoichiometric theoretical equivalent amount, was maintained constant, equal to 1.1 in all the cases. The obtained polyurethane samples were stored at room temperature in a desiccator prior to characterization one week after synthesis. Polyurethane samples were named as PU x:y, x:y being the polyol:BHET-m functional groups equivalent ratio. The polyurethane samples designation, components’ molar ratio, and percentage of components and rerewable carbon content are summarized in Table 1. Figure 1 shows a digital image of the synthesized thermoset polyurethanes.

Thermoset PUs with increasing BHET-m weight content, 0, 8, 12, 17 and 21 wt% from left to right
Chemical Recycling by Glycolysis of the Synthetized Thermoset Polyurethanes
The glycolysis reaction of the synthesized thermoset PUs was carried out in a closed reactor by introducing 30 g of pellets of all the synthesized thermoset polyurethanes containing BHET-m (milled samples from PU 0.8:0.2 to PU 0.5:0.5) and ethylene glycol (EG) in a weight ratio of 4:1 (PU:EG), as described in the literature [33]. Potassium acetate was used as catalyst at a ratio of 0.4 wt% and the reactor was heated to 180 °C for 2 h. It should be noted that a single-phase was observed after the glycolysis process, which consisted mainly on a mixture of polyol-rich and BHET-rich hydroxylated components and the diol used in the glycolysis. After the reaction, the single-phase glycolyzed mixture product was collected and cooled down to room temperature. The glycolyzed mixture product (PU-Gly) was then physico-chemically characterized.
Characterization Techniques
Differential Scanning Calorimetry (DSC)
Thermal properties of BHET-m, polyol and synthesized polyurethane samples, as well as PU-Gly sample were determined by differential scanning calorimetry (DSC, Columbus, Ohio, USA) using a Mettler Toledo DSC3 + device provided with a robotic arm and an electric intracooler as a refrigerator unit. 5–10 mg of sample was encapsulated in aluminium pans and two consecutives dynamic heating scans were performed from − 90 to 240 °C at 20 °C min−1 in nitrogen atmosphere. The glass transition temperature (Tg) of the polyol and the curing enthalpy of the reaction were measured from the first heating scan, as the inflexion point of the curve and the area under the exothermic peaks, respectively. The Tg of the cured thermoset polyurethanes was measured as the inflexion point of the curve of the second heating scan.
Fourier Transform Infrared Spectroscopy (FTIR)
The characteristic functional groups of polyurethanes, BHET-m, polyol and PU-Gly were identified by Fourier transform infrared spectroscopy using a Nicolet Nexus spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) equipped with a MKII Golden Gate accessory (Specac) and a diamond crystal at a nominal incidence angle of 45° and a ZnSe lens. The analyses were carried out in attenuated total reflection (ATR) mode in the 4000–650 cm−1 wavenumber interval, conducting 64 scans with a resolution of 8 cm−1.
Thermogravimetric Analysis (TGA)
The thermal stability of synthesized polyurethanes and recycled BHET was analysed by thermogravimetric analysis (TGA, Columbus, Ohio, USA) using a TGA/SDTA851 Mettler Toledo equipment. Samples were heated from room temperature to 800 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere. In the same way, thermogravimetric study was done for the commercial polyol and glycolyzed PU-Gly sample.
Dynamic Mechanical Analysis (DMA)
The dynamic mechanical behaviour of the thermoset polyurethanes was analysed by DMA in bending mode on an Eplexor Gabo 100N analyser from Netzsch (Selb, Bavaria, Germany). A contact force of 0.8 N, a bending distance of 20 mm and a frequency of 1 Hz were used. Temperature scans were performed from − 80 to 200 °C at a heating rate of 2 °C min−1. Cross-linking density was calculated from the value of storage modulus in the rubbery region according to de Eq. 1 [34].
E’Tα+50 is the storage modulus in the rubbery region (taken at Tα + 50 ºC), R is the universal constant of gases (8.314 J/ (mol ·K)) and Tα is the glass transition temperature in Kelvin taken as the maximum of tan δ peak.
Mechanical Properties
Mechanical flexural tests were carried out using an Instron 5697 equipment (Instron, Norwood, MA, USA) equipped with a 30 kN load cell and with a 3-point bending device. Test were performed at room temperature and at a speed of 1 mm min−1. Samples were cut in 30 mm long, 10 mm wide and 1.5 mm thickness and tested at three-point bending device with a span length of 25 mm. Flexural modulus (E), flexural strength (σ) and strain (ε) were determined from stress–strain curves. In order to determine an average value five different tests were carried out for each sample.
Water Contact Angle (WCA)
The surface hydrophilicity of the different samples was measured at room temperature by static water contact angle (WCA) using the SEO Phoenix Series P-300 equipment (Kromtek Sdn Bhd, Selangor, Malaysia). A deionized water drop of 2 μL was deposited on the material’s surface using a syringe of 0.4 mm diameter and the contact angle value formed by the drop of water was measured ten seconds after the droplet deposition. The average WCA value was obtained for each sample from the result of five water droplets deposited on the polyurethane plaques prepared by compression moulding.
Gel Permeation Chromatography (GPC)
The weight and number average molecular weights, Mw and Mn, respectively, for PU-Gly, bio-polyol and BHET-m samples were determined by gel permeation chromatography (GPC) using a Thermo Scientific chromatograph (Thermo Fisher Scientific, Waltham, Massachusetts, USA) equipped with an isocratic Dionex UltiMate 3000 pump and a RefractoMax 521 refractive index detector. The separation was carried out at 30 °C within four Phenogel GPC columns from Phenomenex with 5 µm particle size and porosities of 105, 103, 100, and 50 Å, located in an UltiMate 3000 thermostated column compartment. Tetrahydrofuran (THF) was used as mobile phase at a flow rate of 1 mL/min. Samples were prepared solving the materials into THF at 1 wt% and filtering with 2 µm pore size nylon filters. Mw and Mn were reported as weight average based on the calibration curve with monodisperse polystyrene standards. THF used in this technique was provided by Macron Fine Chemicals™ (Avantor, Gliwice, Poland).
Results and Discussion
The characterization of the synthesized thermoset polyurethanes was carried out using different techniques. In addition, once the PU plaques were characterized, the chemical recycling of a mixture of all of them was performed to check the recyclability of the thermoset polyurethanes.
Characterization of Synthetized Thermoset Polyurethanes
The curing reaction of the polyurethane mixture was analyzed by dynamic DSC (Fig. 2). In the first heating scan, a Tg was observed for all mixtures, at around − 40 °C, associated to the neat polyol, which shifts to higher temperatures with increasing BHET-m content. Moreover, two exothermic peaks can be observed, the first one around 100 °C, attributed to the curing reaction between pMDI and polyol, as previously reported [35], and the second one around 110 °C, attributed to the curing reaction of the remaining isocyanate groups with BHET-m. It can be observed that the intensity of the second peak increases as does the content of BHET-m.

Dynamic first scan DSC (——) and second scan DSC (–––) thermograms of the different polyurethane systems
The total enthalpy of the curing reaction for each system is summarized in Table 2. As can be seen, the curing enthalpy remains almost constant for the systems containing low BHET-m contents. However, this was not the case for the system with the highest BHET-m content, where a lower enthalpy value was measured.
The higher Tg measured for systems with higher BHET-m content and the lower enthalpy shown by PU 0.5:0:5 could be attributed to part of the curing reaction taking place during the mixing of the components, prior to the DSC test, being this effect more pronounced for increasing BHET-m contents.
In a subsequent dynamic DSC scan, none of the systems showed any trace of residual exothermic heat, regardless the BHET-m content, proving the completion of the curing reaction during the first scan. All systems showed similar Tg values in the second scan, between 80 and 90 °C, proving similar thermal property after a complete curing process.
FTIR spectra of the different synthesized thermoset polyurethanes, together with those of the neat polyol and BHET-m, are presented in Fig. 3. A band characteristic of the isocyanate stretching vibration is observed around 2270 cm−1, which can be attributed to the residual isocyanate (NCO:OH = 1.1) used in excess in the synthesis [16, 36]. However, none of the synthesized polyurethanes showed the characteristic O–H band stretching vibration observed in BHET-m and in polyol at 3342 cm−1 [37], denoting that the polymerization was completed, in agreement with DSC results. In addition, all polyurethane samples showed characteristic bands of urethane groups. A band centered at 3317 cm−1 can be observed, ascribed to stretching vibration of N–H bond [38], which remained constant for all samples due to the similar isocyanate content (Table 1). The bands appearing between 2930 and 2850 cm−1 are related to the absorption bands of the –CH- and –CH2– groups [39]. The band at 1712 cm−1 attributed to the stretching vibration of the carbonyl group encompasses the stretching vibration of the carbonyl group of urethane and also carbonyl of ester observed in the polyol at 1724 cm−1 and in BHET at 1708 cm−1 [40]. Furthermore, a new absorption band is observed for all polyurethanes in the amide II region (1600–1500 cm−1) at 1523 cm−1, related to the bending vibration of N–H combined with the stretching vibration of C–N, characteristic of the urethane group [41, 42]. The bands appearing between 1250 and 1050 cm−1 are related to the CO–O–C asymmetric and symmetric stretching vibrations [43, 44] and remain constant for the synthesized thermoset polyurethanes. Finally, the characteristic band at 725 cm−1, attributed to C–H bond in the aromatic group of BHET, is also observed [13, 45]. The intensity of the band increases with the increase of BHET-m content in the thermoset polyurethane.

FTIR spectra of the synthetized thermoset polyurethane samples, polyol and BHET-m
The thermal stability of the synthetized polyurethanes was analyzed by TGA. The thermograms and the corresponding derivative curves are represented in Fig. 4. For comparison purposes, the thermograms of the neat polyol and BHET-m and their corresponding derivative curves are also represented in Fig. 4. Thermal degradation temperatures are summarized in Table 4. The thermal degradation of the polyol took place between 250 and 474 °C in a two-step weight loss degradation process, as shown by DTG curve where two minimums located at 393 and 470 °C can be observed. It has been reported in the literature that vegetable oil-based polyols usually show a double weight loss [46, 47]. Thermal degradation of PU 1:0, without BHET-m, showed two weight loss steps, the first one, between 300 and 330 °C, was related to the degradation of urethane groups, Td1, and the second one, around 470 °C, to the degradation of the polyol, Td2 [48,49,50]. Concerning the polyurethanes containing BHET-m, it was observed that as the BHET-m content in the thermoset PU increased, the first degradation temperature shifted to lower temperatures, which could be attributed to the lower thermal stability of BHET, which shows a degradation temperature in the 180–280 °C range. Similarly, as summarized in Table 3, the thermoset PU samples containing BHET-m presented higher residue values as the BHET-m content increased. This fact could be due to the incorporation of an aromatic component, such as BHET-m, which generates char during combustion [51].

a TG and b DTG curves of the synthetized thermoset polyurethanes and of the polyol and BHET-m used in the synthesis
The dynamic mechanical behavior of the synthesized thermoset polyurethanes was analyzed in flexural mode. Figure 5 shows the temperature dependence of the storage modulus (E') and loss factor (tan δ). The peak of tan δ is related to the glass transition temperature of thermoset polyurethanes [52]. The Tα values obtained by DMA, together with the values of E' at room temperature and in the rubbery plateau region (at Tα + 50 °C), are summarized in Table 4. The calculated cross-linking density values are also shown in Table 4.

DMA results of the synthetized thermoset polyurethanes
At temperatures below Tα, all systems showed a similar storage modulus, measuring at 25 °C a value around 1.8 GPa for the PU 1:0 and values between 1.8–2.2 GPa for the systems containing BHET-m. However, above Tα, in the rubbery plateau region, lower storage modulus values were measured as BHET-m content increased. In general, although the cross-linking density decreased with increasing BHET-m content, an increase of the Tα values was observed, with the exception of PU 0.5:0.5, for which a lower value was observed. The increase of BHET-m content results in a higher chain mobility restriction, and, consequently, in an increase of the Tα, caused by the steric hindrance of its aromatic structure. The observed trend agrees with the Tg values previously obtained by DSC. Regarding the difference in glass transition temperature values obtained by both techniques, lower Tg values by DSC have been reported in the literature [53]. The mechanical test is much more sensitive than DSC to the changes occurring in this transition, that is why the maximum of tan δ peak is used to determine the glass transition temperature [54, 55]. Regarding cross-linking density, a decrease was observed with the addition of BHET-m, due to replacement of a polyol with functionality higher than 2 by BHET-m with a functionality of 2.
Flexural tests were performed and the stress–strain curves are represented in Fig. 6. Table 5 summarizes the average value of flexural modulus (E), flexural strength (σ) and flexural strain (ε) obtained. It was clearly observed that the addition of BHET-m increased the flexural modulus, whereas, a lower flexural strain was obtained. The aromatic structure of BHET-m provided stiffness to the system [22], however, the incorporation of a difunctional monomer like BHET-m instead of polyol (f = 4) resulted in a lower cross-linking density, as already observed in DMA, resulting in lower strength and earlier breakage of the material, hence, lower flexural strain. Summarizing, the properties of the synthesized thermoset polyurethanes are comparable to those of a biobased PU matrix proposed for composites for structural applications [56].

Mechanical properties of the synthetized thermoset polyurethanes
Finally, the effect of the incorporation of BHET-m on the hydrophilicity of the synthesized polyurethanes was also analyzed. In the synthetized thermoset PU, the density of the hydrophilic dipole-forming urethanes groups [57, 58] remains constant (Table 1), and therefore changes in WCA would be related to the BHET-m content. In this context, as shown in Table 6, an increase in the WCA values was observed with increasing BHET-m content, which can be attributed to the increase in the hydrophobic aromatic content present in the BHET [59]. Hydrophobic surfaces with WCA values of almost 90° were obtained at high BHET-m contents.
Characterization of glycolyzed product from chemical recycling
Chemical recycling processes allow obtaining low molecular weight hydrocarbon basic units, mainly monomers, dimers or oligomers, obtaining high value-added products. The two most successful methods of chemical recycling are glycolysis and hydrolysis, where exchange reactions are used to recover hydroxylated compounds [60]. Regarding PUs, glycolysis has been reported to be the most widely used chemical recycling process [61]. Glycolysis involves an urethane exchange reaction, in which the C-O attached to the carbonyl of the urethane group is exchanged by the hydroxyl group of the glycol, as shown in Fig. 7.

Main glycolysis reaction for polyurethanes
In the synthesized thermoset polyurethanes, the urethane group is linked to the polyol and BHET (Fig. 8), so the exchange could result in a hydroxyl group attached to either the BHET or the polyol rich components, as shown in Fig. 8a and b.

Graphical scheme and reactions that can occur in the glycolysis of synthesized thermoset polyurethanes, a polyol-rich and b BHET-rich hydroxylated components
The glycolyzed mixture product was characterized by several techniques. First, the viscosity values were determined at different temperatures, obtaining 0.8·103, 7.0·103 and 26.7·103 at 50, 35 and 25 ºC, respectively. Moreover, the molecular weight values and the polydispersity were measured by GPC. Figure 9a shows the GPC chromatograms obtained for PU-Gly and for the commercial polyol and the BHET-m used in the synthesis of the thermoset polyurethanes. The glycolyzed mixture product shows a broad size distribution, with elution times ranging from 30 to 41 min that encompasses the distributions observed for the polyol and BHET-m. That is why two intervals have been differentiated in the glycolyzed product, A and B. As represented in Fig. 8b, interval B could be attributed to glycolysis fraction containing BHET-rich monomers, dimers or other alcohols, since the peak observed at 39 min is related to BHET-m and, also, no peak appears in polyol chromatogram at longer retention times. On the other hand, interval A could be attributed mainly to the polyol-rich hydroxylated components represented in Fig. 8a.

a GPC curves of the PU-Gly, and polyol and BHET-m employed in the synthesized thermoset polyurethanes, b FTIR spectra of the glycolyzed product, polyol and thermoset polyurethane, c DSC thermograms of PU-Gly, polyol and synthetized thermoset PU and d) TG (——) and DTG (–––) curves of polyol, BHET-m, synthetized thermoset PU and glycolyzed product.
The weight and number average molecular weights (based on calibration curve with monodisperse polystyrene standards) between elution times of 30.0 and 37.6 min, attributed to polyol-rich fraction, would be Mw = 1475 g/mol and Mn = 936 g/mol, respectively, while between 37.6 and 40.5 min, attributed to BHET-rich fraction, would be Mw = 196 g/mol and Mn = 167 g/mol, respectively. These values are in the range of those measured in Fig. 9a for polyol (Mw = 1379 g/mol and Mn = 1054 g/mol) and for BHET-m (at 37.5 min, Mn = 393 g/mol and Mw = 398 g/mol and at 39.1 min Mn = 168 g/mol and Mw = 174 g/mol). It is worth mentioning that the values measured for BHET-m agree with those previously reported [62, 63]. However, it is important to note that in the interval between 36.8 and 37.9 min, there could be an overlap of peaks pertaining to both the polyol-rich and the BHET-rich fractions. The polyol-rich and BHET-rich fractions, calculated by integrating the area under the peak in each interval, are approximately 72% and 28%, respectively. The obtained BHET-rich fraction was of the order of the BHET-m content average used in the thermoset PU samples, corroborating the occurrence of the glycolysis reaction and the scheme proposed in Fig. 8.
The changes of the functional groups in the PU-Gly with respect to the thermoset polyurethanes synthesized were analyzed by FTIR. Figure 9b shows the spectra of one of the synthesized thermoset polyurethanes, the polyol used in the synthesis and the glycolyzed product. PU-Gly showed a band at 3342 cm−1, which appeared between the absorption band observed in the polyol at 3429 cm−1, characteristic of the –OH stretching vibration, and the absorption band observed in the polyurethane at 3317 cm−1, characteristic of the –NH stretching vibration of the urethane group. Therefore, the absorption band observed in the glycolyzed product suggests that, in addition to the newly formed hydroxyl group, urethane groups remained in the mixture obtained after the glycolysis [41]. The formation of –OH groups during glycolysis was also confirmed by the new absorption bands around 1100–1000 cm−1, which were not present in the polyurethane, and that are characteristic of the absorption of C–OH stretching vibration of primary alcohols [64]. On the other hand, the rest of the bands in the PU-Gly spectrum, such as the stretching vibration of carbonyl groups of esters and urethanes at around 1720–1710 cm−1, combined C–N stretching and N–H bending at 1523 cm−1, C–O stretching vibration at 1220 cm−1 and the aromatic C–H bonds at 725 cm−1, were also observed in the polyurethane. All spectra showed bands related to –CH2– groups around 2950–2850 cm−1 [64].
Thermal properties of the glycolyzed product were analyzed by DSC. The thermograms of PU-Gly, polyol and synthetized PU 0.7:0.3 samples are shown in Fig. 9c. As can be observed, the PU-Gly sample showed a Tg at − 19 °C, which is higher than that of the original polyol, but lower than that of the synthetized PU 07:03. This is attributed to the fact that in the depolymerization process the breakage of the three-dimensional structure occurs randomly, resulting in shorter chains and lower molecular weight, which leads to a lower Tg, due to the higher mobility of the chains compared to the thermoset polyurethane. However, the Tg is higher than that observed at − 58 °C for the starting polyol since the network breakage is not complete, and the chains have more mobility restriction compared to the polyol.
The PU-Gly obtained in the glycolysis of the thermoset polyurethane was also analyzed by TGA. Figure 9d shows the TG and DTG curves of the glycolyzed product, the polyol and BHET used in the synthesis of thermoset polyurethane and the polyurethane itself. Different weight loss steps were observed in the glycolyzed product. The first one around 140–180 °C, with a loss of 4%, could be associated to the EG used in the glycolysis process [65], signaling to some unreacted EG remaining in the single-phase PU-Gly. The second significant weight loss occurs around 300 °C and it was also observed in the thermoset polyurethane sample. This step could be related to the degradation of urethane groups [48, 49], as they were also observed previously by FTIR. Moreover, a shoulder is observed at lower temperatures, around 250 °C, also observed for neat BHET-m, which may be related to the degradation of the BHET-rich components obtained in the glycolyzed product. At higher temperatures, two weight losses were observed, also observed in the polyol at around 350 and 470 °C. It could be concluded that the glycolyzed product is a mixture of different components, some of which are constituted by several functional groups of different thermal stability such as the urethane group or chains derived from vegetable oils, as well BHET. Furthermore, it was observed that the PU-Gly has a significantly high residue, of about 22%. This value is higher than that of the PU 0.7:0.3 sample, and may be due to the fact that all synthetized PU samples were used in the glycolysis, including those with higher BHET-m content, such as PU 0.6:0.4 and PU 0.5:0.5. These samples have higher BHET-m content compared to PU 0.7:0.3, which can generate more char on combustion [51].
Conclusions
In this work, biobased and recycled thermoset polyurethanes have been developed using a castor oil derived polyol and BHET recycled from marine PET litter, producing more environmentally friendly materials. Five different thermoset polyurethanes were synthesized, by varying the ratio of polyol and BHET-m, and characterized in depth to understand the influence of the components on the final properties.
The results showed that marine BHET can be a good alternative for the synthesis of high performance thermoset polyurethanes. DSC and DMA results indicated higher glass transition temperatures for increasing contents of BHET, attributed to the steric hindrance of the aromatic structure of BHET, which restricts the chain mobility. On the other hand, crosslinking densities calculated from DMA results decreased with the replacement of tetrafunctional polyol by difunctional BHET. Furthermore, flexural test results demonstrated that the addition of BHET increases the flexural modulus, leading to stiffer materials, due to the aromatic structure of BHET. The synthesized thermosets showed thermal and mechanical properties comparable to those of PU matrices of composites for structural applications, with PU 0.6:0.4 showing the best properties for this application, and PU 0.8:0.2 presenting still great properties and potential, while showing a high biobased content.
On the other hand, to demonstrate the recyclability of the synthesized thermoset PUs, glycolysis was carried out, from which a single-phase viscous liquid was obtained. The obtained PU-Gly was characterized with several techniques and it was shown that glycolysis resulted in depolymerization of the PUs, resulting in fractions with molecular weights similar to the raw components. Therefore, this preliminary study provides evidence that glycolysis may be a valid strategy for the recycling of the thermoset polyurethanes synthesized in this work.
References
Tournier V, Topham CM, Gilles A, David B, Folgoas C, Moya-Leclair E, Kamionka E, Desrousseaux ML, Texier H, Gavalda S, Cot M, Guémard E, Dalibey M, Nomme J, Cioci G, Barbe S, Chateau M, André I, Duquesne S, Marty A (2020) An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580:216–219. https://doi.org/10.1038/s41586-020-2149-4
Pasula RR, Lim S, Ghadessy FJ, Sana B (2022) The influences of substrates’ physical properties on enzymatic PET hydrolysis: Implications for PET hydrolase engineering. Eng Biol 6:17–22. https://doi.org/10.1049/enb2.12018
Syberg K, Nielsen MB, Westergaard Clausen LP, van Calster G, van Wezel A, Rochman C, Koelmans AA, Cronin R, Pahl S, Hansen SF (2021) Regulation of plastic from a circular economy perspective. Curr Opin Green Sustain Chem. 29:100462. https://doi.org/10.1016/j.cogsc.2021.100462
Nisticò R (2020) Polyethylene terephthalate (PET) in the packaging industry. Polym Test. 90:106707. https://doi.org/10.1016/j.polymertesting.2020.106707
Barnes DKA, Galgani F, Thompson RC, Barlaz M (2009) Accumulation and fragmentation of plastic debris in global environments. Philos Trans R Soc B-Biol Sci 364:1985–1998. https://doi.org/10.1098/rstb.2008.0205
Roager L, Sonnenschein EC (2019) Bacterial candidates for colonization and degradation of marine plastic debris. Environ Sci Technol. https://doi.org/10.1021/acs.est.9b02212
Iñiguez ME, Conesa JA, Soler A (2018) Effect of marine ambient in the production of pollutants from the pyrolysis and combustion of a mixture of plastic materials. Mar Pollut Bull 130:249–257. https://doi.org/10.1016/J.MARPOLBUL.2018.03.040
C. Peña-Rodriguez, G. Mondragon, A. Mendoza, E. Mendiburu-Valor, A. Eceiza, G. Kortaberria, Recycling of marine plastic debris, in: Recent Developments in Plastic Recycling. Springer, 2021: pp. 121–141. https://doi.org/10.1007/978-981-16-3627-1_6.
PlasticsEurope, Plastics—the Facts 2021, (2021). https://plasticseurope.org/knowledge-hub/plastics-the-facts-2021/. Accessed June 10, 2022.
Li B, Wang Z-W, Lin Q-B, Hu C-Y (2016) Study of the migration of stabilizer and plasticizer from polyethylene terephthalate into food simulants. J Chromatogr Sci 54:939–951. https://doi.org/10.1093/chromsci/bmw025
Awaja F, Pavel D (2005) Recycling of PET. Eur Polym J 41:1453–1477. https://doi.org/10.1016/J.EURPOLYMJ.2005.02.005
Mendiburu-Valor E, Mondragon G, González N, Kortaberria G, Martin L, Eceiza A, Peña-Rodriguez C (2022) Valorization of urban and marine PET waste by optimized chemical recycling. Resour Conserv Recycl. 184:106413. https://doi.org/10.1016/j.resconrec.2022.106413
Mendiburu-Valor E, Mondragon G, González N, Kortaberria G, Eceiza A, Peña-Rodriguez C (2021) Improving the efficiency for the production of bis-(2-hydroxyethyl) terephtalate (BHET) from the glycolysis reaction of poly(ethylene terephtalate) (PET) in a pressure reactor. Polymers 13:1461. https://doi.org/10.3390/polym13091461
Hu Y, Wang Y, Zhang X, Qian J, Xing X, Wang X (2020) Synthesis of poly(ethylene terephthalate) based on glycolysis of waste PET fiber. J Macromol Sci Part A 57:430–438. https://doi.org/10.1080/10601325.2019.1709498
George N, Kurian T (2014) Recent developments in the chemical recycling of postconsumer poly(ethylene terephthalate) waste. Ind Eng Chem Res. https://doi.org/10.1021/ie501995m
Maafi EM, Malek F, Tighzert L (2010) Synthesis and characterization of new polyurethane based on polycaprolactone. J Appl Polym Sci 115:3651–3658. https://doi.org/10.1002/app.31448
Yuan Z, Nag R, Cummins E (2022) Ranking of potential hazards from microplastics polymers in the marine environment. J Hazard Mater. 429:128399. https://doi.org/10.1016/j.jhazmat.2022.128399
Singhal P, Small W, Cosgriff-Hernandez E, Maitland DJ, Wilson TS (2014) Low density biodegradable shape memory polyurethane foams for embolic biomedical applications. Acta Biomater 10:67–76. https://doi.org/10.1016/j.actbio.2013.09.027
Kapatel PM, Patel RH (2020) Green approach for the development of novel flame retardant waterborne polyurethanes: synthesis and its characterizations. Mater Today Proc. 23:389–399. https://doi.org/10.1016/j.matpr.2020.02.058
Hao H, Shao J, Deng Y, He S, Luo F, Wu Y, Li J, Tan H, Li J, Fu Q (2016) Synthesis and characterization of biodegradable lysine-based waterborne polyurethane for soft tissue engineering applications. Biomater Sci 4:1682–1690. https://doi.org/10.1039/c6bm00588h
Honarkar H (2018) Waterborne polyurethanes: a review. J Dispers Sci Technol 39:507–516. https://doi.org/10.1080/01932691.2017.1327818
Kopczyńska P, Calvo-Correas T, Eceiza A, Datta J (2016) Synthesis and characterisation of polyurethane elastomers with semi-products obtained from polyurethane recycling. Eur Polym J 85:26–37. https://doi.org/10.1016/j.eurpolymj.2016.09.063
Simón D, Borreguero AM, de Lucas A, Rodríguez JF (2018) Recycling of polyurethanes from laboratory to industry, a journey towards the sustainability. Waste Manage 76:147–171. https://doi.org/10.1016/j.wasman.2018.03.041
Kiss G, Rusu G, Peter F, Tănase I, Bandur G (2020) Recovery of flexible polyurethane foam waste for efficient reuse in industrial formulations. Polymers 12:1533. https://doi.org/10.3390/polym12071533
Gausas L, Kristensen SK, Sun H, Ahrens A, Donslund BS, Lindhardt AT, Skrydstrup T (2021) Catalytic hydrogenation of polyurethanes to base chemicals: from model systems to commercial and end-of-life polyurethane materials. JACS Au 1:517–524. https://doi.org/10.1021/jacsau.1c00050
Pu M, Zhou X, Liu X, Fang C, Wang D (2022) A facile, alternative and sustainable feedstock for transparent polyurethane elastomers from chemical recycling waste PET in high-efficient way. Waste Manage 155:137–145. https://doi.org/10.1016/j.wasman.2022.10.032
Jamdar V, Kathalewar M, Dubey KA, Sabnis A (2017) Recycling of PET wastes using electron beam radiations and preparation of polyurethane coatings using recycled material. Prog Org Coat 107:54–63. https://doi.org/10.1016/j.porgcoat.2017.02.007
Li Q, He H, Zhang C, Liang X, Shen Y (2022) Research on synthesis of polyurethane based on a new chain extender obtained from waste polyethylene terephthalate. J Appl Polym Sci 139:52402. https://doi.org/10.1002/app.52402
Pham CT, Nguyen BT, Nguyen MT, Nguyen TH, Hoang CN, Ngan Nguyen N, Lee P-C, Kim J, Hoang D (2021) The advancement of bis(2-hydroxyethyl)terephthalate recovered from post-consumer poly(ethylene terephthalate) bottles compared to commercial polyol for preparation of high performance polyurethane. J Ind and Eng Chem. 93:196–209. https://doi.org/10.1016/j.jiec.2020.09.024
Cevher D, Sürdem S (2021) Polyurethane adhesive based on polyol monomers BHET and BHETA depolymerised from PET waste. Int J Adhes Adhes. 105:102799. https://doi.org/10.1016/j.ijadhadh.2020.102799
Li M, Luo J, Huang Y, Li X, Yu T, Ge M (2014) Recycling of waste poly(ethylene terephthalate) into flame-retardant rigid polyurethane foams. J Appl Polym Sci. https://doi.org/10.1002/app.40857
Mendiburu-Valor E, Calvo-Correas T, Martin L, Harismendy I, Peña-Rodriguez C, Eceiza A (2023) Synthesis and characterization of sustainable polyurethanes from renewable and recycled feedstocks. J Clean Prod. 400:136749. https://doi.org/10.1016/j.jclepro.2023.136749
Datta J (2010) Synthesis and investigation of glycolysates and obtained polyurethane elastomers. J Elastomer Plast 42:117–127. https://doi.org/10.1177/0095244309354368
Seo J, Yui N, Seo JH (2019) Development of a supramolecular accelerator simultaneously to increase the cross-linking density and ductility of an epoxy resin. Chem Eng J 356:303–311. https://doi.org/10.1016/j.cej.2018.09.020
Echeverria-Altuna O, Ollo O, Larraza I, Gabilondo N, Harismendy I, Eceiza A (2022) Effect of the biobased polyols chemical structure on high performance thermoset polyurethane properties. Polymer. https://doi.org/10.1016/j.polymer.2022.125515
Pan X, Webster DC (2012) New biobased high functionality polyols and their use in polyurethane coatings. Chemsuschem 5:419–429. https://doi.org/10.1002/cssc.201100415
Siroèiae AP, Fijaèko A, Hrnjak-Murgiae Z (2013) Chemical recycling of postconsumer poly(ethylene-terephthalate) bottles-depolymerization study. Chem Biochem Eng Q 27:65–71
Calvo-Correas T, Santamaria-Echart A, Saralegi A, Martin L, Valea Á, Corcuera MA, Eceiza A (2015) Thermally-responsive biopolyurethanes from a biobased diisocyanate. Eur Polym J 70:173–185. https://doi.org/10.1016/J.EURPOLYMJ.2015.07.022
Da X, Liu C, Long Y, Xie X (2020) Polyurethane foaming with CO2 adducts from C8 alkyl grafted polyethyleneimines: Optimization of the grafting rate and application of the blowing agents. J Appl Polym Sci 137:48752. https://doi.org/10.1002/app.48752
Ryszkowska JL, Auguścik M, Sheikh A, Boccaccini AR (2010) Biodegradable polyurethane composite scaffolds containing Bioglass® for bone tissue engineering. Compos Sci Technol 70:1894–1908. https://doi.org/10.1016/J.COMPSCITECH.2010.05.011
Calvo-Correas T, Ugarte L, Trzebiatowska PJ, Sanzberro R, Datta J, Corcuera MÁ, Eceiza A (2017) Thermoplastic polyurethanes with glycolysate intermediates from polyurethane waste recycling. Polym Degrad Stab 144:411–419. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2017.09.001
Datta J, Głowińska E (2014) Effect of hydroxylated soybean oil and bio-based propanediol on the structure and thermal properties of synthesized bio-polyurethanes. Ind Crops Prod 61:84–91. https://doi.org/10.1016/J.INDCROP.2014.06.050
Tang Q, Gao K (2017) Structure analysis of polyether-based thermoplastic polyurethane elastomers by FTIR, 1H NMR and 13C NMR. Int J Polym Anal Charact 22:569–574. https://doi.org/10.1080/1023666X.2017.1312754
Pérez-Limiñana MA, Arán-Aís F, Torró-Palau AM, Orgilés-Barceló AC, Martín-Martínez JM (2005) Characterization of waterborne polyurethane adhesives containing different amounts of ionic groups. Int J Adhes Adhes 25:507–517. https://doi.org/10.1016/j.ijadhadh.2005.02.002
Bhattacharyya A, Mukherjee D, Mishra R, Kundu PP (2017) Preparation of polyurethane–alginate/chitosan core shell nanoparticles for the purpose of oral insulin delivery. Eur Polym J 92:294–313. https://doi.org/10.1016/j.eurpolymj.2017.05.015
Jayavani S, Sunanda S, Varghese TO, Nayak SK (2017) Synthesis and characterizations of sustainable polyester polyols from non-edible vegetable oils: Thermal and structural evaluation. J Clean Prod 162:795–805. https://doi.org/10.1016/j.jclepro.2017.06.040
Zhang M, Pan H, Zhang L, Hu L, Zhou Y (2014) Study of the mechanical, thermal properties and flame retardancy of rigid polyurethane foams prepared from modified castor-oil-based polyols. Ind Crops Prod 59:135–143. https://doi.org/10.1016/j.indcrop.2014.05.016
Corcuera MA, Rueda L, Saralegui A, Martín Ma.D., B. Fernández-d’Arlas, I. Mondragon, A. Eceiza, (2011) Effect of diisocyanate structure on the properties and microstructure of polyurethanes based on polyols derived from renewable resources. J Appl Polym Sci. 122:3677–3685. https://doi.org/10.1002/app.34781
Bueno-Ferrer C, Hablot E, M. del C. Garrigós, S. Bocchini, L. Averous, A. Jiménez, (2012) Polym Degrad Stab. 97:1964–1969. https://doi.org/10.1016/j.polymdegradstab.2012.03.002
Gonella LB, Zattera AJ, Zeni M, Oliveira RVB, Canto LB (2009) New reclaiming process of thermoset polyurethane foam and blending with polyamide-12 and thermoplastic polyurethane. J Elastomer Plast 41:303–322. https://doi.org/10.1177/0095244309099413
Lee PS, Jung SMG (2022) Flame retardancy of polyurethane foams prepared from green polyols with flame retardants. J Appl Polym Sci 139:52010. https://doi.org/10.1002/app.52010
Achorn PJ, Ferrillot RC (1994) Comparison of thermal techniques for glass transition measurements of polystyrene and cross-linked acrylic polyurethane films. J Appl Polym Sci 54:2033–2044
Zhang J, Zhang C, Madbouly SA (2015) In situ polymerization of bio-based thermosetting polyurethane/graphene oxide nanocomposites. J Appl Polym Sci 132:41751. https://doi.org/10.1002/app.41751
J.C. Domínguez, Rheology and curing process of thermosets, in: Thermosets: Structure, Properties, and Applications: Second Edition, Elsevier, 2018: pp. 115–146. https://doi.org/10.1016/B978-0-08-101021-1.00004-6.
Menard KP, Menard NR (2020) Dynamic mechanical analysis. CRC Press. https://doi.org/10.1201/9780429190308
Echeverria-Altuna O, Ollo O, Larraza I, Elizetxea C, Harismendy I, Eceiza A (2022) Development of a Novel Biobased Polyurethane Resin System for Structural Composites. Polymers. https://doi.org/10.3390/polym14214553
B. Fernández-D’Arlas, A. Alonso-Varona, T. Palomares, M.A. Corcuera, A. Eceiza, (2015) Studies on the morphology, properties and biocompatibility of aliphatic diisocyanate-polycarbonate polyurethanes. Polym Degrad Stab. 122:153–160. https://doi.org/10.1016/j.polymdegradstab.2015.10.023
B. Fernémdez-D’Arlas, L. Rueda, K. de La Caba, I. Mondragon, A. Eceiza (2008) Microdomain composition and properties differences of biodegradable polyurethanes based on MDI and HDI, Polym Eng Sci. 48: 519–529. https://doi.org/10.1002/pen.20983.
Sarkar K, Rama S, Meka K, Bagchi A, Krishna NS, Ramachandra SG, Madras G, Chatterjee K (2014) Polyester derived from recycled poly(ethylene terephthalate) waste for regenerative medicine. RSC Adv. https://doi.org/10.1039/c4ra09560j
Schneiderman DK, Vanderlaan ME, Mannion AM, Panthani TR, Batiste DC, Wang JZ, Bates FS, Macosko CW, Hillmyer MA (2016) Chemically recyclable biobased polyurethanes. ACS Macro Lett 5:515–518. https://doi.org/10.1021/acsmacrolett.6b00193
Wu C-H, Chang C-Y, Li J-K (2002) Glycolysis of rigid polyurethane from waste refrigerators. Polym Degrad Stab 75:413–421. https://doi.org/10.1016/S0141-3910(01)00237-3
López-Fonseca R, Duque-Ingunza I, de Rivas B, Flores-Giraldo L, Gutiérrez-Ortiz JI (2011) Kinetics of catalytic glycolysis of PET wastes with sodium carbonate, Chem. Eng J 168:312–320. https://doi.org/10.1016/J.CEJ.2011.01.031
Fang P, Liu B, Xu J, Zhou Q, Zhang S, Ma J, X. lu, (2018) High-efficiency glycolysis of poly(ethylene terephthalate) by sandwich-structure polyoxometalate catalyst with two active sites. Polym Degrad Stab. 156:22–31. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2018.07.004
Ştefănescu M, Stoia M, Ştefănescu O, Davidescu C, Vlase G, Sfîrloagǎ P (2010) Synthesis and characterization of poly (vinyl alcohol)/ethylene glycol/silica hybrids. Thermal analysis and FT-IR study. Rev Roum Chim. 55:17–23
Pasha A, Khasim S, Al-Hartomy OA, Lakshmi M, Manjunatha KG (2018) Highly sensitive ethylene glycol-doped PEDOT-PSS organic thin films for LPG sensing. RSC Adv 8:18074–18083. https://doi.org/10.1039/c8ra01061g
Acknowledgements
Financial support from the Basque Country Government in the framework of Grupos Consolidados (IT-1690-22) and through the ELKARTEK 2021 (Project NEOMAT KK-2021/00059) programme is gratefully acknowledged. The University of the Basque Country (UPV/EHU) (GIU18/216 Research Group) is also gratefully acknowledged. The authors would like to thank the Circular Economy University-Company Classroom (Faculty of Engineering Gipuzkoa, UPV/EHU, Provincial Council of Gipuzkoa). We are also grateful to the Macrobehavior-Mesostructure-Nanotechnology SGIker unit of UPV/EHU. E. M.-V. would like to thank the Basque Government for the PhD grant (PRE_2018_1_0014).
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. Eusko Jaurlaritza, PRE_2018_1_0014, ELKARTEK, NEOMAT KK-2021/00059, Euskal Herriko Unibertsitatea, GIU18/216 Research Group.
Author information
Authors and Affiliations
Contributions
Author Contributions: Eider Mendiburu-Valor (E.M.-V.): Methodology, Investigation, Writing, Visualization, Formal analysis. Izaskun Larraza (I.L.): Methodology, Investigation, Writing. Oihane Echeverria-Altuna (O.E-A): Methodology, Investigation. Isabel Harismendy (I.H): Methodology, Investigation. Cristina Peña-Rodriguez (C.P.-R.): Conceptualization, Methodology, Supervision, Writing, Visualization, Formal analysis, Funding acquisition. Arantxa Eceiza (A.E.): Conceptualization, Methodology, Supervision, Writing, Visualization, Formal analysis, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mendiburu-Valor, E., Larraza, I., Echeverria-Altuna, O. et al. Thermoset polyurethanes from biobased and recycled components. J Polym Environ 31, 4946–4959 (2023). https://doi.org/10.1007/s10924-023-02891-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-023-02891-1