
Abstract
In this work rigid polyurethane foams (PUR) were obtained by replacement of 0–70 wt% of petrochemical polyol with bio-polyol obtained via cellulose liquefaction in presence of crude glycerol. The foams with different content of a bio-polyol were prepared by single step method for NCO/OH ratio equals 1.5. The prepared materials were analyzed in terms of their morphology, chemical structure, thermal stability and basic physical and mechanical properties. The effects of photo-oxidative and thermo-oxidative aging on chemical structure, apparent density and mechanical properties of the biomass based rigid polyurethane foams were investigated and discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polyurethanes are among the most commonly used plastics. They are obtained through polyaddition of diisocyanates and polyols. Diversity of their properties enables their application in various branches of industry, e.g. automotive industry or production of thermal insulation materials [1]. Majority of commercially available polyurethanes are produced from petroleum-based materials. Over the last years, utilization of renewable materials, such as lignocellulose biomass, is a very popular topic among the researchers all over the world [2,3,4,5,6]. It is related to the various law regulations, ongoing trends associated with the “green chemistry” and diminishing resources of petroleum [7, 8].
Liquefaction of biomass with organic solvents (e.g. multi-hydroxyl group alcohols) at elevated temperatures is based on the solvolysis process, which results in decomposition of chemical bonds present in biomass [5]. Such process leads to generations of liquid products, e.g. bio-polyols, which can be subsequently incorporated into production of eco-friendly PUR materials [9,10,11]. Various sources of biomass were investigated for production of bio-polyols, such as corn bran [11], rice straw [12], wheat straw [12], bagasse, cotton stalks [13], wood [14], rapeseed cake, apple pomace [15] and algae [16]. Prepared bio-polyols were used to obtain bio-PUR, with properties often comparable to materials obtained from petroleum-based polyols [17,18,19].
Very important for the efficiency of liquefaction process and properties of final product is selection of proper solvent, which is used in excess [20]. It can increase the cost of bio-polyol production and have negative impact on future commercialization of such technologies. Interesting alternative for petrochemical solvents can be crude glycerol. It is a by-product of biodiesel production, for each tonne of biofuel, around 100 kg of glycerol is generated [21]. Crude glycerol contains various impurities, such as methanol, soaps, free fatty acids and residual catalysts [22]. In the literature there are various reports on crude glycerol utilization in production of hydrogen, 1,3-propanediol, succinic acid or polyhydroxyalkanoates [23, 24]. Other interesting application is biomass liquefaction.
Hu et al. [25] described liquefaction of soybean straw with crude glycerol. They determined optimal process parameters for production of bio-polyols − 240 °C, > 180 min, 3 wt% of H2SO4 as catalyst and 10–15 wt% of biomass. Bio-polyols were characterized by hydroxyl value from 440 to 540 mg KOH/g, acid value lower than 5 mg KOH/g and viscosity from 16 to 45 Pa·s. Subsequently, the authors used obtained bio-polyol for preparation of rigid PUR foams, which showed apparent density from 33 to 37 kg/m3 and compressive strength from 148 to 227 kPa. Such results indicate that crude glycerol can be successfully applied as an alternative solvent in lignocellulose biomass liquefaction process resulting in bio-polyols production.
An important parameter in the use of rigid polyurethane foams is their resistance to aging. The useful life of polymers for outdoor applications is highly dependent on atmospheric conditions, which often cause a decrease in physical properties [26]. The temperature of the initial urethane bond breakdown depends on the structure of the isocyanate (NCO) and the polyol used [27]. For example, the polyurethanes obtained from aromatic isocyanates will turn yellow when exposed to UV radiation. This is due to the oxidation reaction in the polymer backbone [28]. In addition to radiation, the presence of oxygen is an important parameter as it speeds up the aging process of polyurethane materials, eg the initial thermal decomposition temperature in the air is about 50 °C lower than in N2 [29].
In presented research work, crude glycerol-based bio-polyol described in the previous study [30] was used to prepare rigid polyurethane foams. Its content in polyol mixture was altered from 0 to 70 wt%. Processing, physical (sol fraction), chemical (FTIR studies) and cellular (microscopic analysis) structure, mechanical (compression tests, dynamic mechanical analysis) and thermal (thermogravimetric analysis) properties of resulting rigid foams were examined. The effects of the content of bio-polyol in foams’ formulations on photo-oxidative, thermo-oxidative aging were examined.
Experimental
Materials
Bio-polyol (EP1) obtained via cellulose liquefaction with crude glycerol described in previous work [30] was used to prepare rigid PUR foams. It was characterized by hydroxyl value (LOH) of 754 mg KOH/g, a water content of 0.63% and viscosity of 7.5 Pa·s (at 25 °C). Bio-polyol was used together with commercially available polyol from PCC Rokita company—oligoether Rokopol RF55, characterized by molecular weight approximately 550 g/mol and LOH ca. 495 mg KOH/g, water content of 0.1 wt% and viscosity of 9.2 Pa·s (at 25 °C). Isocyanate used in the reaction was polymeric 4,4′-methylene diphenyl diisocyanate (pMDI) with 31.5% content of NCO groups (BASF). The average functionality of pMDI was c.a. 2.8. Tin(II) 2-ethylhexanoate (K12) produced by Sigma Aldrich, tertiary amine Dabco® TMR-2 and Dabco® 1027 produced by Air Products Europe Chemicals B.V. were used as catalysts. TEGOSTAB B 8465 produced by Evonik Industries AG was used as a stabilizer of porous structure. Carbon dioxide generated in the reaction of water and isocyanate groups was a chemical blowing agent. Additional physical blowing agent was a Solkane® 365/227 produced by Solvay.
Preparation of Rigid Polyurethane Foams
Rigid polyurethane foams were produced on a laboratory scale by a single step method from a two-component (A and B) system with the ratio of NCO/OH groups equals to 1.5. The component A (polyol mixture) consisting of the proper amounts of Rokopol RF 55 or a mixture of Rokopol RF55 and the bio-based polyol EP1 in an appropriate ratio, catalysts, surfactant, physical blowing agent and water was weighed and placed in a 500 ml polypropylene cup. Such prepared component A was mixed with component B (polyisocyanate, pMDI) at a predetermined mass ratio and stirred at 3000 rpm for 15 s. The resulting reaction mixture was poured into an open metal mould of dimensions 20 × 20 cm. After demoulding, the obtained PUR samples were kept at 60 °C for 24 h and then seasoned at room temperature for 24 h. Table 1 contains the details of foam formulations.
Photo-Oxidative Aging of Rigid PUR Foams
Foams were subjected to photo-oxidative aging due to ultraviolet radiation (UV-A) according to EN 927-6:2007 standard. During test, cylindrical samples with dimensions of 18 × 20 mm were irradiated for 300 h. Samples were irradiated with UV-A 340 lamps, λ = 360 nm, which accounts for energy of ~ 330 kJ/mol. Such energy is enough to cause dissociation of bonds present in polyurethane backbone and generate free radicals initiating decomposition processes. Aging was performed in air atmosphere at constant temperature of 23 °C and relative humidity of 60%.
Thermo-Oxidative Aging of Rigid PUR Foams
Foams were subjected to thermo-oxidative aging in accordance to ASTM D3574-03 standard. Cylindrical samples with dimensions of 18 × 20 mm were kept at 140 °C for 22 h. After that, samples were cooled down before further analyses.
Characterization of Rigid PUR Foams
The following process parameters were observed and characterized: start time (time elapsed from the start of the process until the start of volume expansion); rise time (time elapsed until the foam reaches its maximum height); and gel time (time elapsed until the surface of the foam stops being tacky to the touch).
The soluble (sol) fraction of each sample was determined by Soxhlet extraction, using xylene as a solvent. Around 1.5 g of sample was put inside a cellulose thimble and submitted to an extraction process for 24 h. After extraction, the samples were dried for 24 h at 80 °C to remove the solvent and their weight was measured. The sol fraction was determined as the ratio of the difference between weight of the sample before extraction (W1) and weight of the sample after extraction (W2), relative to the weight of the sample before extraction (W1), according to Eq. (1):
FT-IR spectrophotometric analysis was performed in order to examine the structure of the bio-based polyol and rigid PUR foams. The analysis was performed at a resolution of 4 cm− 1 using a Nicolet 8700 apparatus (Thermo Electron Corporation) equipped with a snap-Gold State II which allows for making measurements in the reflection configuration mode.
The apparent density of polyurethanes samples was calculated in accordance to PN–EN ISO 845:2000, as the ratio of the sample weight to the sample volume. The volume of the samples having a cubic shape was measured with a slide caliper having an accuracy of 0.1 mm. The samples were weighed using an electronic analytical balance with an accuracy of 0.1 mg.
The compression strength of the PUR samples was estimated in accordance to PN–EN ISO 604:2006. The samples of cubic shape and dimensions of 50 × 50 × 50 mm3 were measured with a slide caliper with an accuracy of 0.1 mm. The compression test was performed on a Zwick/Roell tensile tester at a constant speed of 10 mm/min to 40% deformation.
The thermogravimetric analysis (TGA) was performer on a NETZSCH TG 209 apparatus using 5 mg samples within the temperature range 100–600 °C under nitrogen atmosphere, at a heating rate of 15 °C/min.
Dynamic mechanical analysis was performed using DMA Q800 TA Instruments apparatus. Samples were analyzed in compression mode with a frequency of 1 Hz. Measurements were performed for the temperature range from 25 to 270 °C with heating rate 4 °C/min. Samples were cylindrical-shaped with dimensions of 8 × 12 mm.
The cell morphology of polyurethane samples was investigated with a Philips-FEI XL 30 environmental scanning electron microscope (SEM) using an acceleration of 25 kV. The samples were cut at room temperature, while the observations were performed in wet mode.
Results and Discussion
Figure 1 shows the effect of addition of bio-polyol to the PUR foam formulation on processing times and synthesis temperature. Incorporation of prepared bio-polyol into formulation of prepared foams did not result in noticeable changes of processing times. Such phenomenon can be considered beneficial from the technological point of view, because the changes of formulation do not require special adjustement of apparatus’ settings. Moreover, introduction of bio-polyol into the formulation led to the increase in temperature during the process of foaming PUR rigid foams. This was due to a higher reactivity of the modified polyol system compared to the pure petrochemical Rokopol 55.

Effect of bio-polyol content on processing times (a) and temperature (b) of PUR foam synthesis
In Table 2 there are presented properties of rigid PUR foams. Despite the slight increase of apparent density, all analyzed foams, independently from bio-polyol content, showed similar value of apparent density on the level of 41 ± 2 kg/m3. Authors of other works often observed noticeable increase of apparent density, while increasing share of bio-polyols from liquefaction process in polyol mixture [31]. Maintaining the apparent density of obtained materials at the same level can be considered very beneficial, because it does not require the incorporation of higher amounts of foaming agents. Such phenomenon can be associated with the presence of residual lower molecular weight compounds in obtained bio-polyol, which are volatile in temperatures reached during manufacturing of rigid PUR foams.
In Table 2 there are also presented parameters describing foams’ cellular structure, such as cell size, cell aspect ratio and cell roundness. All parameters were determined from SEM images of samples presented in the Fig. 2, using ImageJ computer software. In order to calculate shape descriptors software fitted ellipses into foams’ cells and used the following formulas (2 and 3):
where L l and L s mean the length of longer and shorter axis of fitted ellipse and A is the area of ellipse.

SEM images of prepared PUR samples
Modification of foams’ formulation with 70 wt% of crude glycerol-based bio-polyol resulted in the noticeable, 25% decrease of average cell size in comparison to the reference sample, which can be considered as very beneficial for potential application of this material as thermal insulation. Similar values of cell size were reported for PUR foams used traditionally for insulation of district heating pipes [32]. Similar effects associated with the use of crude glycerol-based bio-polyols in rigid PUR foams were previously observed [33]. Changes in cellular structure can be also related to the lower viscosity of applied bio-polyol in comparison to Rokopol RF55, which enabled generation of more uniform structure.
Aspect ratio and roundness are equal to 1 in case of perfectly circular pores. In case of analyzed foams, the rise of temperature during synthesis could indicate the increase of cells’ anisotropy, due to the faster evaporation of foaming agents. Nevertheless, it can be seen that the increase of bio-polyol content up to 47 wt% resulted in the increase of cells’ homogeneity. Such phenomenon can be associated with relatively high values of polyols’ density.
In Table 2 there are also presented values of sol fraction of PUR foams determined during swelling test. The increase of bio-polyol’s share in polyol mixture reduced the sol fraction at almost 3 times, which may point to the increase of cross-link density, suggested previously by the analysis of temperature during foams’ preparation. Similar results were observed by other researchers [34].
Generally, mechanical performance of polyurethane foams, as well as other porous materials, are strongly related to their apparent density, which determines the share of solid material in its volume. During compression, distribution of external force inside the material is changing due to the changes of foams’ cellular structure. Higher apparent density is obviously related to higher structural packaging, which additionally enhances the stiffness of material [35]. In the Fig. 3 there are presented values of compressive strength of analyzed PUR samples. Incorporation of bio-polyol into foams’ formulation resulted in the increase of foams’ mechanical performance. Such enhancement could be associated with the rise of crosslink density of material, suggested by swelling tests, as well as, more uniform cellular structure of foams containing more bio-polyol in formulation [35]. Compression tests were conducted in two directions: parallel and perpendicular to the foaming direction, in order to determine the influence of cellular structure on mechanical performance. Data shown in the Fig. 3 points to correlation between structural and mechanical anisotropy. Such difference in compressive performance in these two directions may be related to the aspect ratio of cells present in the structure and their elongation in the foam’s rise direction. Similar observations related to the structural and mechanical anisotropy were noted by other researchers [36, 37].

Effect of bio-polyol content on compressive strength
In the Fig. 4 there are shown FTIR spectra of prepared rigid PUR foams. Absorption bands, characteristic for the stretching vibrations of N–H bonds were observed in the range of 3290–3320 cm− 1, while signals attributed to their bending vibrations were observed at 1510–1520 cm− 1 [38]. In the range of 1705–1715 cm− 1, peaks related to the stretching vibrations of C=O bonds were noted. Signals around 1200–1215 cm− 1 are associated with the stretching of C–N bonds present in urethane groups [39]. All signals mentioned above confirm the presence of urethane groups in analyzed samples.

FTIR spectra of prepared polyurethane foams
Absorption bands observed at 2860–2870 and 2960–2975 cm− 1 can be attributed to the symmetric and asymmetric stretching vibrations of C–H bonds in methylene and methyl groups present in polymer backbone. Low intensity peaks observed at 2260–2280 cm− 1 are related to the presence of N=C=O groups, due to the excess of isocyanate used during preparation of rigid PUR foams (high isocyanate index) [40]. Signals in the range of 1410–1415 cm− 1 are associated with the vibrations of isocyanurate rings, products of isocyanate groups’ trimerization, which are very important for thermal stability of rigid PUR foams [41]. Slight increase of the intensity of these signals may suggest enhancement of thermal stability, which was further confirmed by TGA analysis. Relatively weak peak around 1310 cm− 1 is related to the vibrations of N–H and C–N bonds [42]. Multiplet signals in the range of 1000–1090 cm− 1 are related to the vibrations of δ bonds between carbon and oxygen atoms (in ester and ether groups), related to the chemical structure of applied polyols [43].
Polyurethane foams are considered to be thermally unstable due to the presence of urethane bonds which, depending on the isocyanates and polyols used, begin to degrade between 150 and 220 °C [44]. As shown in Table 3, introduction of bio-polyol into the formulation of rigid PUR foams resulted in the shift of onset of thermal degradation (determined as 2 wt% mass loss of foam) towards higher temperature. Depending on formulation, thermal degradation starts in the range of 200–250 °C. This can be attributed to relatively unstable urethane bonds and/or pyranose rings [45, 46]. The second stage of degradation that occurred between about 350 and 490 °C can be attributed to the degradation of the polyol components. The intensity of this peak is significantly lower than of the peak at 350 °C, which means that the rate of degradation of the rigid segments is higher [47, 48]. At about 475 °C there is the thermolysis of organic residues from the previous degradation stages [49]. Replacement of petrochemical polyol with the bio-polyol resulted in peak shifts in the DTG curves (Fig. 5), which confirms the changes in thermal stability of foams. The rigid polyurethane foams obtained in this work are characterized by thermal stability comparable to others obtained from liquefied biomass. For example, Zhao et al. [45], have prepared polyurethane foam obtained from the solubilization of the bark of the pineapple. The foams were characterized by two-stage degradation, which occurred at about 250–400 °C, respectively.

Differential thermogravimetric curves for investigated polyurethane foams
Photo-Oxidative and Thermo-Oxidative Aging
In the Fig. 6 there are presented FTIR spectra of rigid PUR foams subjected to photo-oxidative and thermo-oxidative aging in order to determine the influence of these processes on the chemical structure of analyzed materials. Obtained results are similar to those described by other researchers [50]. Both methods of aging result in the significant decrease of the intensity or even disappearance of the signal attributed to the vibrations of N=C=O groups around 2260–2280 cm− 1. Such effect can be related to the chemical reactions involving these groups, supported by the UV radiation and elevated temperature [51]. Moreover, after aging of foams, noticeable differences in the shape and intensity of signals around 3290–3320 cm− 1 (stretching vibrations of N–H bonds in urethane groups) can be observed, which points to decomposition of urethane groups. Such phenomenon was confirmed by other researchers [52]. More significant changes in FTIR spectra were observed in case of foams subjected to photo-oxidation. Generally, it can be seen that intensity of majority of signals is decreasing. A band at 1170 cm− 1 appears, which is attributed, among others, to bridging between polyol segments [53]. The decrease of the intensity of signal around 1310 cm− 1 suggests the conversion of urethane bond into ortho-aromatic amine ester structure via photo-Fries rearrangement [54]. In the range of 1705–1715 cm− 1, lowering of peaks’ intensity is observed, which is related to the UV-induced oxidation of C=O. The absorption bands characteristic for the aromatic ring (1510, 1410 cm− 1) are also reduced, showing that aromatic structures in polyurethanes are unstable in UV radiation [51].

FTIR spectra of rigid polyurethane foams prior to aging and after aging
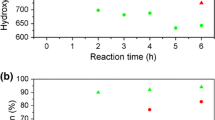
The accelerated aging process was also used to better understand the effect of the degradation process of rigid polyurethane foams on mechanical properties and apparent density. Larger differences in compressive strength and apparent density were observed for foams aged at 140 °C compared with UV-aged foams. This is due to the well-known effect of expanding polyurethane foams with simultaneous loss of weight after heating. Thermal degradation includes physical processes (volatile compounds release) and chemical (chain cleavage, oxidation, depolymerization) [50]. Figure 7 shows the compressive strength values at 20% deformation and the apparent density for aged foams and non-aged foams. For UV-aged foams a slight decrease in apparent density is observed by approx. 1% on average, and for thermally aged foams, the apparent density decreases by an average of about 4%. As the apparent density decreases, there is a decrease in compressive strength. It has been found that aging with UV radiation does not significantly affect the compressive strength of the tested materials. Similar dependencies have been observed by others [55, 56]. With increasing bio-polyol content in the formulation of rigid polyurethane foams larger differences in apparent density and compressive strength are observed. This is confirmed by the analysis of FTIR spectra, where greater changes in the shape of the peaks connected with urethane bonds in foams with the higher bio- polyol content were observed..

Comparison of apparent density and compressive strength of rigid polyurethane foams before and after aging
Conclusions
The presented results demonstrate that the introduction of bio-polyols, were prepared via cellulose liquefaction process, into the structure of rigid polyurethane foams allows for obtaining materials with properties comparable to the petrochemical foams. The foams produced with the use of biomass based polyols were characterized by slightly increased apparent density and simultaneously noticeably enhanced compressive strength, which is connected to the differences in chemical structure of bio-polyols and petrochemical polyols. The SEM photos of surface of rigid polyurethane foams showed decrease of cell size with the addition of bio-polyol and changes in cell aspect ratio and cell roundness. Replacement of the petrochemical polyol by bio-polyol resulted in lowering of thermal stability of foams. The impact of thermo-oxidative and photo-oxidative aging on the structure and properties of rigid polyurethane foams was also investigated. The apparent density and mechanical properties of bio-polyol based PUR decreased obviously with aging process time. Bigger changes in the apparent density and compressive strength were observed for thermo-oxidative aging. In summary, this work has confirmed that biomass based polyols can be successfully used in the polyurethane foam production. Foams produced with the use of obtained bio-polyols are characterized with acceptable mechanical and their properties, comparable to those prepared with petrochemical polyols. Moreover, their preparation can be performed in favorable conditions and with reduced costs, compared to unmodified materials.
References
Akindoyo JO, Beg MdDH, Ghazali S, Islam MR, Jeyaratnam N, Yuvaraj AR (2016) RSC Adv 6:114453–114482
Gandini A (2008) Macromolecules 41:9491–9504
Jo YJ, Choi SH, Lee EY (2013) Appl Chem Eng 24:579–586
Jo YJ, Ly HV, Kim J, Kim SS, Lee EY (2015) J Ind Eng Chem 29:24–31
Hu S, Luo X, Li Y (2014) ChemSusChem 7:66–72
Gama NV, Soares B, Freire CSR, Silva R, Brandäo I, Neto CP, Barros-Timmons A, Ferreira A (2015) Polym Int 64:250–257
Eissen M, .Metzger JO, Schmidt E, Schneidewing U (2002) Angew Chem Int Ed 41:414–436
Bhunia HP, Nando GB, Chaki TK, Basak A, LenKa S, Nayak PL (1999) Eur Polym J 35:1381–1391
Xu J, Jiang J, Hse CY, Shupe TF (2014) J Appl Polym Sci 131:1–7
Kurimoto Y, Takeda M, Koizumi A, Yamauchi S, Doi S, Tamura Y (2000) Bioresour Technol 74:151–157
Lee SH, Yoshioka M, Shiraishi N (2000) J Appl Polym Sci 78:319–325
Liang L, Mao Z, Li Y, Wan C, Wang T, Zhang L, Zhang L (2006) Bioresources 1:248–256
Hassan EBM, Shukry N (2008) Ind Crop Prod 27:33–38
Kurimoto Y, Koizumi A, Doi S, Yamauchi S, Ono H (2001) Biomass Bioenerg 21:381–390
Briones R, Serrano L, Labidi J (2012) J Chem Technol Biotechnol 87:244–249
Kim KH, Jo YJ, Lee ChG, Lee EY (2015) Algal Res 12:539–544
Zhang HR, Pang H, Shi JZ (2012) J Appl Polym Sci 123:850–856
Zhang HR, Ding F, Luo CR, Xiong L, Chen XD (2012) Ind Crop Prod 39:47–51
Soares B, Gama N, Freire C, Barros-Timmons A, Brandão I, Silva R, Ferreira A (2014) ACS Sustain Chem Eng 2:846–854
Chen FG, Lu ZM (2009) J Appl Polym Sci 111:508–516
Johnson DT, Taconi KA (2007) Environ Prog 26:338–348
Thompson JC, He BB (2006) Appl Eng Agric 22:261–265
Dobroth ZT, Hu SJ, Coats ER, McDonald AG (2011) Bioresour Technol 102:3352–3359
Pachauri N, He B (2006) In Presented at 2006 ASABE Annual International Meeting, paper number: 066223
Hu S, Wan C, Li Y (2012) Bioresour Technol 103:227–233
Davis A (1977) In: Grassie N (ed) The weathering of polymers in development of polymer degradation. vol 1. Applied Science Publishers, London
Chattopadhyay D, Webster D (2009) Prog Polym Sci 34:1068–1133
Rosu D, Rosu L, Cascaval CN (2009) Polym Degrad Stab 94:591–596
Jiao L, Xiao H, Wang Q, Sun J (2013) Polym Degrad Stab 98:2687–2696
Kosmela P, Hejna A, Formela K, Haponiuk JT (2016) Ł Piszczyk Cellulose 23:2929–2942
Lu X, Wang Y, Zhang Y, Cheng X, Yu Y, Jin Y (2016) J Wuhan Univ Technol 31:918–924
Jarfelt U, Ramnas O (2006) 10th Internationl Symposium on District Heating and Cooling, 3–5 Sep 2006
Hejna A, Kirpluks M, Kosmela P, Cabulis U, Haponiuk JT, Piszczyk Ł (2017) Ind Crop Prod 95:113–125
Wang T, Li D, Wang L, Yin J, Chen XD, Mao Z (2008) Chem Eng Res Des 86:416–421
Mosiewicki MA, Dell’Arciprete GA, Aranguren MI, Marcovich NE (2009) J Compos Mater 43:3057–3072
Kurańska M, Prociak A, Kirpluks M, Cabulis U (2015) Ind Crop Prod 74:849–857
Modesti M, Lorenzetti A (2003) Eur Polym J 39:263–268
Sormana JL, Meredith JC (2004) Macromolecules 37:2186–2195
Fournier D, Du F, Prez (2008) Macromolecules 41:4622–4630
Jiao L, Xiao H, Wang Q, Sun J (2013) Polym Degrad Stabil 98:2687–2696
Samborska-Skowron R, Balas A (2003) Polimery 48:371–374
Silverstein RM, Webster FX, Klemie DJ (2005) Spectrometric identification of organic compounds, 7th edn, Willey, New York
Pretsch T, Jakob I, Müller W (2009) Polym Degrad Stabil 94:61–73
Lee SH, Teramoto Y, Shiraishi N (2002) J Appl Polym Sci 83:1482–1489
Zhao Y, Yan N, Feng M (2012) J Appl Polym Sci 123:2849–2858
Hakim AAA, Nassar M, Emam A, Sultan M (2011) Mater Chem Phys 129:301–307
Cervantes-Uc JM, Moo Espinosa JI, Cauich-Rodriguez JV, Avila-Ortega A, Vazquez-Torres H, Marcos-Fernandez A (2009) J San Roman Polym Degrad Stab 94:1666–1677
Pawlik H, Prociak A (2012) J Polym Environ, 20:438–445
Zhang L, Zhang M, Zhou Y, Hu L (2013) Polym Degrad Stabil, 98:2784–2794
Yarahmadi N, Vega A, Jakubowicz I (2017) Polym Degrad Stabil 138:192–200
Paberza A, Stiebra L, Cabulis U (2015) J Renew Mater 3:19–27
Romanova V, Begishev V, Karmanov V, Maitz MF (2002) J Raman Spectrosc 33:769–777
Cipriani E, Bracco P, Kurtz SM, Costa L, Zanetti M (2013) Polym Degrad Stab 98:1225–1235
Gardette JL, Lemaire J (1984) Polym Degrad Stabil 6:135–148
Yarahmadi N, Sällström JH (2014) Improved maintenance strategies for district heating pipe lines, 14th International Symposium on District Heating and Cooling, September 7–9, 2014, Stockholm, Sweden
Leuteritz A, Döring. K-D, Lampke T, Kuehnert I (2016) Polym Test 51:142–147
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kosmela, P., Hejna, A., Formela, K. et al. The Study on Application of Biopolyols Obtained by Cellulose Biomass Liquefaction Performed with Crude Glycerol for the Synthesis of Rigid Polyurethane Foams. J Polym Environ 26, 2546–2554 (2018). https://doi.org/10.1007/s10924-017-1145-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-017-1145-8