
Abstract
The aliphatic polyurethanes based on atactic poly[(R,S)-3-hydroxybutyrate] (a-PHB) and commercial oligomerols: poly(ε-caprolactone)diol and polyoxytetramethylenediol were investigated. a-PHB was obtained by anionic ring-opening polymerization of (R,S)-β-butyrolactone. The 4,4′-methylenedicyclohexyl diisocyanate and 1,4-butanediol were used as contributors of hard segments. The aim of the study was to determine the influence of synthetic, atactic a-PHB in soft segments of polyurethanes on their degradability in simulated body fluids (SBF) and Ringer solution. The incubation of polymer samples in both degradative solutions was carried out for 36 weeks. It was concluded that the presence of a-PHB in polyurethane structure accelerated their degradation in SBF and in Ringer solution and, protected the calcification process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polyurethanes are commonly used in medicine as degradable and non-degradable implants [1, 2]. The unique combination of their properties as flexibility, toughness and biocompatibility makes them a proper material for medical engineering. For the tissue regeneration process the physico-mechanical properties of the implanted material must be similar to the surrounding natural tissue. If required, the implant should also degrade at a rate similar to the native tissue regeneration process. The biodegradability of polyurethanes can be accelerated by using appropriate substrates for their creation.
Polyesterols such as poly(ε-caprolactone)diol (PCL) and polyetheroles such as polyoxytetramethylenediol (PTMG) can be incorporated into soft segments (SSs) of polyurethanes. PCL is a biocompatible, biodegradable and semi-crystalline aliphatic polyester, well known for possessing a valuable set of properties, such as nontoxicity for living organisms and resorption after an appropriate period of implantation time. It undergoes the enzymatic degradation and slow hydrolytical processes. The hydrolysis of the ester linkage is the main mechanism of PCL biodegradation [3]. Whereas PTMG oligoetherol, used as SSs of polyurethanes, degrades mainly by oxidizing mechanisms [4].
Atactic poly[(R,S)-3-hydroxybutyrate] (a-PHB), the synthetic amorphous analog of natural polyhydroxybutyrate, is known as a biodegradable and biocompatible polymer [5, 6] and as a OH-terminated polyol can also be incorporated into polyurethane structure. Previous work showed that the presence of a-PHB in polyurethane network influences their structural, thermal, mechanical and degradation properties [7–10]. Polyurethanes based on a-PHB do not affect the blood parameters in direct contact and seem to be biostatic against pathogenic microorganisms [10–12].
Furthermore, application of aliphatic diisocyanate for hard segment creation (instead of aromatic one) prevents the risk of aromatic diamine formation as a by-product of the polyurethane degradation.
We report in this current study on the influence of synthetic, atactic a-PHB used as a part of a SS of aliphatic polyurethanes on their degradability in simulated body fluid (SBF) and in Ringer solution. The rate of degradation was monitored by the weight loss of samples during incubation in solutions by the microscopic observations of sample surfaces and by evaluating its thermal properties using differential scanning calorimetry (DSC).
Experimental
Materials
The samples studied were obtained at our laboratories as described previously [7, 13]. Synthesis of polyurethanes was carried out in a two-step reaction, with molar ratio of NCO:OH = 3.7:1 or 2:1, respectively, at prepolymerization stage.
Hard segments of all polyurethanes were made from 4,4′-methylenedicyclohexyl diisocyanate (H12MDI) (Alfa Aesar) and 1,4-butanediol (1,4-BD) (Aldrich). SSs were formed from the mixture of synthetic a-PHB (Mn 2000) and PTMG (Mn 2000, Aldrich) or from a-PHB and PCL (Mn 1920, Aldrich). a-PHB was obtained by anionic ring-opening polymerization of (R,S)-β-butyrolactone initiated by 3-hydroxybutyric acid sodium salt/18-crown-6 complex at room temperature and terminated with 2-bromoethanol [14].
Eight kinds of polyurethanes were studied. Two of them contained synthetic a-PHB and PTMG in SSs, with 14 and 17.5 % of a-PHB in polymer bulk for PURPTMG+a-PHB/3.7 and PURPTMG+a-PHB/2 respectively. SSs of the next two polyurethanes were built from a-PHB and PCL, also with 14 and 17.5 % of a-PHB in polyurethane bulk for PURPCL+a-PHB/3.7 and PURPCL+a-PHB/2 respectively. Polyurethanes without a-PHB in SSs were investigated for comparison.
Investigated polyurethanes showed low-molecular masses (Mn ranging from 7,900 to 18,400) and dispersity (Mw/Mn ranging from 2.6 to 3.95) [9]. As it was described before, polyurethanes were hydrogen-bonded, and for samples that contained PTMG in SSs the creation of a small amount of allophanate structures was observed [9]. The wetting angle of polyurethanes based on a-PHB was ranged from 57.5° to 64.6°, which has indicated on their hydrophility. It may therefore be expected that their hydrophility can facilitate their degradation [15].
Degradation in Simulated Body Fluid
Simulated body fluid was prepared as follows: firstly 8.035 g NaCl, 0.355 g NaHCO3, 0.225 g KCl, 0.231 g K2HPO4·3H2O, 0.311 g MgCl2·6H2O and 39 mL of 1 M hydrochloric acid were dissolved into 750 mL of deionized water (UV sterilized), then 0.292 g CaCl2, 0.072 g Na2SO4 and 6.118 g tris(hydroxymethyl) aminomethane were slowly added in turn. Finally the fluid was buffered to 1 L at physiological pH 7.4 at 37 °C with dilute hydrochloric acid solution [16].
In vitro degradation in SBF was carried out for 4, 12, 24 and 36 weeks by immersing weighted polyurethanes samples in freshly prepared SBF. The medium was refreshed every week.
Degradation in Ringer Balanced Salt Solution
Ringer solution was prepared by dissolving four tablets (Merck) in 500 mL of deionized and UV sterilized water with sodium azide (0.02 %). Composition of one tablet was: 0.00525 g NH4Cl, 0.005 g NaHCO3, 0.04 g CaCl2·2H2O, 0.00525 g KCl and 1.125 g NaCl. Incubation of polymer samples was carried out at 37 °C for 4, 12, 24 and 36 weeks. The incubating solution was replaced every month [17].
The Weight Changes
The polyurethanes were cut before incubation into samples with an area about 1 cm2, dried to a constant weight at 50 °C in vacuum drier, and weighed using an analytical balance. Next, they were immersed in sterilized containers and covered with appropriate solution using 10 mL per square of polymer sample and incubated in heating chamber in 37 ± 1 °C.
The samples were removed from each degradative solution at the given time, rinsed three times with distilled water, dried to a constant weight at 50 °C in vacuum drier and weighed to determine the weight change. The presented values of experimental weight changes were the arithmetic mean of three measurements.
Differential Scanning Calorimetry
Changes in thermal properties of polyurethane samples after their incubation in SBF and Ringer solutions were determined using of TA-DSC 2010 apparatus (TA Instruments, Newcastle, DE, USA). The instrument was calibrated with high purity indium. The degraded polyurethane samples were dried to constant weight before DSC measurement. The specimens were heated in sealed aluminium pans and scanned from −80 to 200 °C (first run) at heating rate of 10 °C/min, rapidly cooled to −80 °C and heated again to 200 °C (second run). All experiments were carried out under nitrogen atmosphere (flow of 50 mL/min). The melting temperature (Tm) of the specimen was taken as the peak temperature maximum of the melting endotherm from first run, and the glass transition temperature (Tg) was taken as the midpoint of the increase of the specific heat associated with the transition (second run). In case of polyurethanes obtained with NCO:OH = 3.7:1 and degraded in both solutions, the heating run was performed only to 160 °C because of their thermal degradation above that temperature.
Microscopic Observation
Microscopic observation of polymer surface changes during degradation was performed in reflected light with an optical microscope Nicon Alphaphot-2YS2 connected with digital photo camera Casio QY2900UX, at magnification 1:300.
Results and Discussion
Polyurethanes with various SSs and length of hard segments were studied and their compositions are summarized in Table 1.
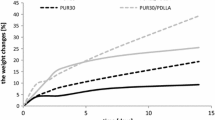
The weight changes of polyurethane samples after incubation in SBF and in Ringer solution are presented in Figs. 1 and 2 respectively.

The weight changes of polyether-esterurethane (a) and polyesterurethane (b) samples during incubation in SBF

The weight changes of polyether-ester-urethane (a) and polyesterurethane (b) samples during incubation in Ringer solution
The mass changes of polymer samples with a-PHB after incubation in SBF indicated that polyurethanes with high content of hard segments (obtained at NCO:OH = 3.7:1) were degraded faster than polyurethanes with lower molar ratio of NCO:OH in prepolymer (Fig. 1). It was observed both for both: polyether-esterurethane (Fig. 1a) and polyesterurethane (Fig. 1b). It could be expected that the higher amount of ester groups in the structure of PURs (obtained at NCO:OH = 2:1 in prepolymer) may facilitate the hydrolysis of PURPTMG+a-PHB/2 and PURPCL+a-PHB/2 as compared with PURPTMG+a-PHB/3.7 and PURPTMG+PCL/3.7 respectively. Moreover, the higher content of hard segments should cause the slower hydrolytic degradation of polyurethanes [18, 19]. However, taking into consideration the generally lower density of our polyurethanes obtained at NCO:OH = 3.7:1 compared to NCO:OH = 2:1, in prepolymer step [15], the incubated medium with inorganic salts could easy diffuse into polymers network, which was observed. Only low decrease or even increase of samples mass after 36 weeks of incubation in SBF was indicated (Fig. 1).
In our previous study we demonstrated, that the decrease of molecular mass of all investigated polyurethanes after hydrolysis in phosphate buffer took place, which indicated on macrochains scission but only small weight loss of polymer samples was noticed [15].
Introducing of atactic a-PHB into the polymer structure accelerated the degradation of polyurethanes in SBF. It was especially visible in case of polyurethanes with higher content of hard segments. Polyurethane with PTMG and a-PHB in SSs—PURPTMG+a-PHB/3.7—appeared as the most susceptible to SBF degradation from the all investigated polymers. It may be supposed that the water penetration into the polymer network followed by the hydrolysis of macrochains was facilitated due to more amorphous morphology of this polyurethane (Tables 3, 4, 5, 6). Because of the presence of ether groups in SSs (not susceptible to hydrolysis) this polyurethane degrades mostly via chemical breakage of the urethane bonds joining hard and SSs [20].
Previous hydrolytic degradation studies showed that PURPTMG+a-PHB/3.7 underwent the hydrolysis after 36 weeks of incubation in phosphate buffer. The sample mass was reduced by about 61 % [8]. However, the weight loss of polyurethane samples after the degradation in SBF was lower than in the case of degradation in mentioned phosphate buffer. It may be due to the fact that the molecules of salts were trapped between the macrochains of the polymer network and influenced on the samples weight.
Observed for PURs without a-PHB negligible decrease or even small increase of sample weight at the end of incubation could be explained by solid salt confined in polymer network.
The microscope images indicated that the presence of a-PHB in SSs appeared to protect polyurethanes against the salt sediments (Figs. 3, 4).

Microscopic surface changes of polyetherurethane and polyether-esterurethane before (“0”) and after 36 weeks of incubation in SBF

Microscopic surface changes of polyesterurethane before and after 36 weeks of incubation in SBF
It was also observed that the sediment of solid salt on the surface of the polymer was larger after the incubation in SBF for polyurethanes synthesized at NCO:OH = 3.7:1 than at NCO:OH = 2:1 (in prepolymer step), what may indicate on their higher tendency to calcify. It could be caused by the generally higher hydrophobicity of PUR with molar ratio of NCO:OH = 2:1 in prepolymer [15]. The calcification process could be also connected with the structure of microchains. In case of higher amount of hard segments more binding sites for the formation of calcific nuclei were probably presented.
The polyurethanes synthesized at NCO:OH = 3.7:1 and susceptible to salt sediment were also more susceptible to degradation than synthesized at NCO:OH = 2:1 (in prepolymer step). Their surface was scored and cracked with black areas (Figs. 3, 4).
The same tendency as in case of incubation in SBF was observed for weight changes during the degradation in Ringer solution (Fig. 2).
The lower density of polyurethanes contained PTMG in SSs (in comparison to that obtained from PCL) caused, that degradative solutions could easy diffuse into the polymer structure [15].
The analytical data (glass temperature Tg of SSs, Tm and their enthalpy ΔH) from DSC curves of polyurethanes before and after degradation are summarized in Tables 3 and 5 for polyether-esterurethane and in Tables 4 and 6 for polyester-urethane. Thermal properties of oligomerols are presented in Table 2, for comparison.
The glass temperature Tg of SSs of obtained polyurethanes (Tables 3, 4, 5, 6) was higher than the Tg of a-PHB, PCL and PTMG monomers. This result indicated that the soft phase was influenced by the urethane hard segments, resulting in phase mixing. It may suggest the low phase separation, especially in case of polyurethanes based on PCL and a-PHB. Similar results were reported by Korley et al. [21], where Tg of SSs of aliphatic polyurethanes only slightly increased from that of pure polyol, what was explained as a relatively small amount of hard segments mixing within the soft domain.
The investigated polyurethanes were only slightly crystalline (Tables 3, 4, 5, 6). For initial PURPCL+a-PHB/2 a crystalline phase of a SS was observed on DSC curves as the endothermic peak with maximum at 70.1 °C and with 26.2 J/g of melting enthalpy. In case of PURPCL+a-PHB/3.7 where the content of SSs was lower, the arrangement of PCL was very difficult and crystals could not be created.
During incubation in both solutions some macrochains were cut, then reorganized and some small degree of crystallinity disappeared (e.g. PURPTMG+a-PHB/3.7 and PURPCL+a-PHB/3.7) whereas another—appeared (e.g. PURPTMG+a-PHB/2 and PURPCL+a-PHB/2). As it was reported by Hong et al. [22], during the incubation of polyurethane based on poly(3-caprolactone-co-β-butyrolactone) in hydrolytic solution, molecules of water diffused into the amorphous region and the degradation of polymer chains was easier. In consequence the mobility of chains increased which facilitates their crystallization.
The high exothermic peak on DSC thermograms with maximum in range of 164–177 °C and enthalpy even up to 95 J/g for PURPTMG+a-PHB/3.7 and PURPCL+a-PHB/3.7 (degraded in both solutions) were observed. It indicated on high rate of degradation process of these polyurethanes, what was in agreement with observed weight loss (Fig. 1). The similar exothermic processes were not observed for PURs with lower NCO:OH ratio.
Different changes in polyurethanes structure after incubation in both solutions studied were observed, depending on SSs structure and molar ratio of NCO:OH in prepolymer step.
Noticed for PURPTMG+a-PHB/2 and PURPCL+a-PHB/2 after the incubation in SBF (Tables 3, 4) and in Ringer solution (Table 6) decreasing of Tg was probably the result of increasing of chains mobility after their scission [23].
However, for polyurethanes obtained at NCO:OH = 3.7:1 in prepolymer and degraded both in SBF and in Ringer solutions (Tables 3, 4, 5, 6) Tg shifted to the higher temperatures in comparison to that of non degraded PURs samples. This could be explained by considerable structural changes connected with the reduction of chain lengths followed by eventual physical crosslinking of macromolecules after degradation [18, 24].
Generally it could be stated, that more significant changes of thermal properties observed after degradation of polyurethanes synthesized with higher content of hard segments confirmed their higher degradability.
Conclusion
The results of this study revealed that the polyurethanes synthesized at molar ratio of NCO:OH = 3.7:1 appeared more susceptible to degradation in SBF and Ringer balanced salt solution as well as to salt sedimentation when compared with polyurethanes obtained at NCO:OH = 2:1 (in prepolymer step). Thermal properties observed after degradation of polyurethanes with higher content of hard segments also confirmed their higher degradability.
The presence of a-PHB in the structure of investigated polyurethanes accelerated their degradation in both solutions studied, protected them against the calcification process and affected their morphology. Polyurethane with PTMG and a-PHB in SSs and synthesized at NCO:OH = 3.7:1 in prepolimer step was the most susceptible to degradation in simulated body fluids.
It could be stated that polyurethanes based on atactic a-PHB displayed the properties appropriate for further investigations for medical application as e.g. degradable scaffolds.
References
Efe T, Getgood A, Schofer MD, Fuchs-Winkelmann S, Mann D, Paletta JRJ, Heyse TJ (2012) Knee Surg Sports Traumatol Arthrosc 20:1822
Mândru M, Ciobanu C, Vlad S, Butnaru M, Lebrun L, Popa M (2013) Cent Eur J Chem 11(4):542
Mondal S, Martin D (2012) Polym Degrad Stab 97:1553
Santerre JP, Woodhouse K, Laroche G, Labow RS (2005) Biomaterials 26:7457
Zawidlak-Węgrzyńska B, Kawalec M, Bosek I, Łuczyk-Juzwa M et al (2010) Eur J Med Chem 45:1833
Piddubnyak V, Kurcok P, Matuszowicz A, Głowala M, Fiszer-Kierzkowska A, Jedliński Z, Juzwa M, Krawczyk Z (2004) Biomaterials 25:5271
Brzeska J, Dacko P, Janeczek H, Kowalczuk M, Janik H, Rutkowska M (2011) Polimery 56(1):27
Brzeska J, Dacko P, Heimowska A, Janik H, Kowalczuk M, Rutkowska M (2012) Ochrona przed korozją 1:8
Brzeska J, Dacko P, Gębarowska K, Janik H, Kaczmarczyk B, Kasperczyk J, Kowalczuk M, Rutkowska M (2012) J Appl Polym Sci 125(6):4285
Brzeska J, Kowalczuk M, Janik H, Rutkowska M (2012) Zeszyty naukowe Akademii Morskiej w Gdyni. Jt Proc 74:5
Brzeska J, Janik H, Kowalczuk M, Rutkowska M (2011) Eng Biomater 65(XIV):106–108
Brzeska J, Janik H, Kowalczuk M, Rutkowska M (2011) Eng Biomater 73(XIV):106–108
Brzeska J, Dacko P, Janik H, Kowalczuk M, Rutkowska M (2012) Biodegradowalne poliuretany i sposób ich wytwarzania, PL Patent Number 212763
Arslan H, Adamus G, Hazer B, Kowalczuk M (1999) Rapid Commun Mass Spectrom 13:2433
Brzeska J (2010) Thesis Gdynia Maritime University
Kokubo T, Takadama H (2006) Biomaterials 27:2907
Wan Y, Wen D (2005) J Membr Sci 246:193
Wojturska J (2011) Polimery 56(3):177
Umare SS, Chandure AS (2008) Chem Eng J 142:65
Hepburn C (1992) Polyurethane elastomers. Elsevier, London
Korley LTJ, Pate BD, Thomas EL, Hammond PT (2006) Polymer 47:3073
Hong JH, Jeon HJ, Yoo JH, Yu W-R, Youk JH (2007) Polym Degrad Stab 92:1186
Feng Y, Li C (2006) Polym Degrad Stab 91(8):1711
Ryszkowska J, Auguścik M, Sheikh A, Boccaccini AR (2010) Compos Sci Technol 70:1894
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Brzeska, J., Heimowska, A., Janeczek, H. et al. Polyurethanes Based on Atactic Poly[(R,S)-3-hydroxybutyrate]: Preliminary Degradation Studies in Simulated Body Fluids. J Polym Environ 22, 176–182 (2014). https://doi.org/10.1007/s10924-014-0650-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-014-0650-2