
Abstract
We study single-voxel in vivo proton magnetic resonance spectroscopy (MRS) of white matter in the brain of a 25 year old healthy male volunteer. The free induction decay (FID) data of short length (0.5KB) are encoded at a long echo time (272 ms) with and without water suppression at a clinical scanner of a weak magnetic field (1.5T). For these FIDs, the fast Fourier transform (FFT) gives sparse, rough and metabolically uninformative spectra. In such spectra, resolution and signal to noise ratio (SNR) are poor. Exponential or Gaussian filters applied to the FIDs can improve SNR in the FFT spectra, but only at the expense of the worsened resolution. This impacts adversely on in vivo MRS for which both resolution and SNR of spectra need to be very good or excellent, without necessarily resorting to stronger magnetic fields. Such a long sought goal is at last within reach by means of the optimized derivative fast Fourier transform (dFFT), which dramatically outperforms the FFT in every facet of signal estimations. The optimized dFFT simultaneously improves resolution and SNR in derivative spectra. They are presently shown to be of comparably high quality irrespective of whether water is suppressed or not in the course of FID encodings. The ensuing benefits of utmost relevance in the clinic include a substantial shortening of the patient examination time. The implied significantly better cost-effectiveness should make in vivo MRS at low-field clinical scanners (1.5T) more affordable to ever larger circles of hospitals worldwide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
One of the most powerful strategies for structure determinations of any molecule is nuclear magnetic resonance (NMR) spectroscopy [1,2,3,4,5,6,7,8]. In medical diagnostics, this non-ionizing and non-invasive methodology, synonymously called magnetic resonance spectroscopy (MRS), can detect molecular changes prior to their manifestations on anatomical images to aid the disease control and eventual cure [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41]. Therein, data analysis and interpretation depend critically on reliable processing of time signals that are alternatively referred to as free induction decay (FID) data in the form of curves [42,43,44,45,46,47,48,49,50].
In MRS and throughout the multi-disciplinary field of signal processing, the frequently employed shape estimator is the fast Fourier transform (FFT), either in its unweighted (unattenuated) or weighted (attenuated) versions [51, 52]. Abundant usage of the FFT occurs despite its low resolution and poor signal-to-noise ratio (SNR) [11, 45, 46]. Such severe drawbacks should be surmountable by another shape estimator, the unattenuated derivative fast Fourier transform (dFFT). However, this is not the case for noisy time signals (measured or synthesized alike) [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68].
In fact, quite the opposite occurs for noise-corrupted FIDs for which the dFFT becomes inferior even to the already inadequate FFT regarding both resolution and SNR. This is attributed to multiplication of the given time signal c(t) by a power function \(t^m\) (\(0\le t\le T,\, m=1, 2, 3,\ldots \)). The latter monomial is produced by applying the \(m\,\)th derivative operator \((\text{ d }/\text{d }\nu )^m\) to the given continuous FFT spectrum \(F(\nu )\). Here, T is the total acquisition time of c(t) and \(\nu \) is the linear frequency.
In the FFT, frequencies \(\nu \) are fixed in advance (predetermined exclusively by T). They are artificial as they do not correspond to the characteristic frequencies from which the given FID is built. The mentioned power function \(t^m\) amplifies noise at larger values of time t. This is exacerbated by the fact that encoded FIDs are dominated by noise at larger t (i.e. at their tails). Therefore, the dFFT is bound to fail, as it processes directly the unweighted product \(t^mc(t)\).
The FFT is model-independent as it does not assume any mathematical function for time signals nor spectral lineshapes. Such a critically important asset in signal processing is inherited by the dFFT, but the expected advantage is left unexploited due to the mentioned breakdown of the latter estimator. Moreover, the computational expedience of the FFT through the fast algorithm of Cooley and Tukey [69] is preserved by the dFFT, but this is of no practical significance either because of the unacceptable derivative Fourier spectra. Hence, for noise-contaminated FIDs, it would be very important to regularize the divergence features of the dFFT so that it could synchronously ameliorate resolution and SNR, while retaining the model-independence and the fast Cooley-Tukey computations.
For measured FIDs, resolution and SNR exhibit a dual uncertainty (indeterminacy) relationship. The implication is that, particularly for these time signals of most interest in practice, attempts to simultaneously improve resolution and SNR are predicted to fail. Such restrictions amply hold true in the FFT and are much more exacerbated in the dFFT for measured time signals [53,54,55,56,57,58,59,60,61,62,63,64,65,66]. This is a further motivation to rescue the situation by a judicious regularization via e.g. a suitable optimization procedure applied to the dFFT.
The sought regularization, yielding the optimized dFFT, has recently been implemented for FIDs encoded by in vitro MRS [67, 68]. This is achieved through countering the adverse effects of the monomial \(t^m\) by multiplying it with a decaying filter \(\textrm{e}^{-\lambda _p(m,T)t^p}\), which is an exponential (\(p=1\)) or a Gaussian (\(p=2\)), where \(\lambda _p(m,T)>0\). The goal of the ensuing product \(t^m\textrm{e}^{-\lambda _p(m,T)t^p}\) is to strike a balance between the derivative order m and the sought resolution for the amount of noise contained in the encoded FID. As a result of this trade-off, the optimized dFFT can simultaneously increase both resolution and SNR to a sufficient level needed in practice for vastly different applications.
In order to optimize resolution and SNR at the same time, these two attenuators, the exponential filter (EF) and the Gaussian filter (GF), are adapted to the derivative order m from the time power function \(t^m\). Appropriately then, the product \(t^m\textrm{e}^{-\lambda _p(m,T)t^p}\) is called the adaptive power-exponential filter (APEF) for \(p=1\) and the adaptive power-Gaussian filter (APGF) for \(p=2\). Thus, the unattenuated dFFT and the optimized dFFT share the same monomial \(t^m\), but it is only the latter processor that additionally includes the adaptive damping function \(\textrm{e}^{-\lambda _p(m,T)t^p}\). The key advantages of the optimized dFFT should be beneficial to the entire signal processing field with versatile applications in physics, chemistry, medicine and elsewhere.
The optimized dFFT can alternatively be conceived as a special version of the attenuated FFT, which represents the customary FFT applied to the given FID weighted with the APEF or APGF. This helps appreciate that the optimized dFFT can be built directly into the magnetic resonance instruments (NMR spectrometers, clinical scanners for MRS) by allowing the user of the FFT to opt for either the APEF or APGF as the special weight function \(w_p(t)=\textrm{e}^{-\lambda _p(m,T)t^p}\) which multiplies the encoded FID.
While viewing the optimized dFFT as this specially attenuated FFT, weighted by the APEF or APGF, might be convenient in practice, it is nevertheless important to always bear in mind the true origin of the monomial \(t^m\). The origin of \(t^m\) in the APEF or APGF is in applying the multi-derivative operator \((\text{ d }/\text{d }\nu )^m\) to the harmonic function \(\textrm{e}^{-2\pi it\nu }\) from the continuous Fourier transform. This remark (backed by the unsmoothing property of derivatives) points to the reason for which \(t^m\) by itself leads to simultaneous lineshape narrowing (resolution improvement) and peak intensity enhancing for noiseless FIDs.
The underlying mechanism for the favorable effect of the multi-derivatives \((\text{ d }/\text{d }\nu )^m\) is best appreciated for chemical shifts away from the given \(k\,\)th fundamental resonance frequency \(\nu _k\). Namely, from both sides of the center of a resonance, the \(m\,\)th derivative lineshape decays faster as \(1/\nu ^{m+1}_k\, (m\ge 1)\) than \(1/\nu _k\) in the FFT [55]. This yields the peak base contraction which, in turn, suppresses the overlap of the adjacent resonances.
This lineshape tightening or localizing leads to the peak width decreasing and the concomitant peak height increasing. The opposite occurs in the FFT lineshapes that fall off too slowly (\(\sim 1/\nu _k\), as stated) and thus exhibit far extending tails prone to partially or completely mask the neighboring and even distant peaks. This increases the likelihood for overlaps of resonances. However, the powerful action of \(t^m\) is annulled by noise, which always contaminates encoded FIDs. In such cases, the damping function \(\textrm{e}^{-\lambda _p(m,T)t^p}\) needs to multiply the time monomial to minimize noise amplification by \(t^m\) at larger t. In other words, the action of \(\textrm{e}^{-\lambda _p(m,T)t^p}\) is to temper a bad or diverging behavior of \(t^mc(t)\) at the tail of the given measured c(t).
Recently [67], the optimized dFFT has been applied to the FIDs encoded by in vitro proton MRS at a clinical scanner of low static magnetic field strength (\(B_0=1.5\)T) from a Philips phantom [70] as well as at a 600 MHz (\(B_0\approx 14.1\)T) Bruker spectrometer from a specimen of human malignant ovarian cyst fluid [71]. The short FIDs (0.5KB, total signal length \(N=512\)) for the said phantom [70] have been encoded at a long echo time (TE=272 ms) with as well as without water suppression, and these FIDs were subjected to the optimized dFFT using the APEF and APGF, respectively [67].
The resulting spectra reconstructed by the optimized dFFT for the water-suppressed and water-unsuppressed FIDs were of a comparable excellent quality, regarding the three pillars of proper derivative spectroscopy [67]:
-
(i) high resolution with narrowest full widths at half maximae (FWHM),
-
(ii) maximized SNR at all resonance frequencies,
-
(iii) steady lineshapes for increasing m with no derivative-induced artifacts.
In Ref. [71], water has been suppressed in the process of encoding the long FIDs (16 KB, \(N=16384\)) at short echo time (TE=30 ms) from a sampled biofluid (malignant ovarian cyst) of a patient. In this case, the optimized dFFT with the APEF also showed an excellent performance by bringing over a dozen of the well-resolved peaks close to the chemical shift axis in an exemplified narrow frequency band around the dominant lactate doublet resonance (a recognized cancer biomarker) [67]. This took place despite an enormous dynamic range of spectral intensities, judged upon the peak height ratio of the order of about \(6\times 10^3\) between the tallest and the shortest resonances (6041/0.85 for lactate/isoleucine).
In our follow-up study [68], some further instructive insights into the optimized dFFT with the APEF were gained on the strongly overlapped resonances, corresponding to the mentioned FID from a patient [71]. Therein, highlighted were the chemical shift bands in the aliphatic region containing the choline compounds (recognized cancer biomarkers) and the citrate multiplets (potential cancer biomarkers). In particular, it was found that an unassigned resonance, invisibly glued to free choline in the FFT envelope, becomes sharply separated as a well-delineated peak in the optimized dFFT spectrum.
Failing to detect this closely adjacent structure would yield about twice the true peak area as well as the concentration of free choline. This error would impact adversely on the diagnostic accuracy in distinguishing between the benign and cancerous cases. Such an example shows that the optimized dFFT can peer into the possible compositeness of even an apparently symmetric singlet from the FFT (e.g. free choline).
The moral of this story is that each resonance should, in principle, be treated as a potential multiplet, prior to eventually uncovering its physical constituents as e.g. purely true singlets, doublets, triplets, quartets, etc. In human biofluids and tissues scanned by MRS, tightly overlapped peaks abound because of the J-coupling of nuclei, as well as due to the fact that many metabolites have similar intrinsic spin-spin relaxation times \(T_2\). Moreover, these metabolites resonate at close chemical shifts under the same acquisition conditions.
The present analysis furthers the optimized dFFT with the help of the APEF and APGF by focusing on the following aspects:
-
Derivation of the analytical forms of the damping parameters \(\lambda _p(m,T)\) for the optimization by highlighting the origin of the simultaneous adaptation to the derivative order m and SNR of the FID.
-
Reconstructions of high-resolution and low-noise spectra by utilizing short FIDs (0.5 KB) encoded using a low-field clinical scanner (1.5T) by in vivo proton MRS at a long echo time (272 ms) with and without water suppression from white matter in the brain of a 25 year old healthy male volunteer. In pursuing this work, the related earlier studies [67, 68] on the optimized dFFT are complemented by the present main goal for encoded FIDs:
-
Verifying whether the performance of the optimized dFFT for these two drastically different in vivo FIDs could be of a similar high quality as that for the mentioned in vitro FIDs [67] under the same acquisition conditions: \(B_0=1.5\)T, total signal length \(N=512\) (0.5KB), 128 transients to be averaged for the SNR amelioration, bandwidth BW = 1000 Hz, repetition time TR = 2000 ms and echo time TE = 272 ms.
2 Theory
2.1 Fast Fourier transform
For a time signal c(t) or FID, given as a continuous function of independent variable t in the interval [0, T], the finite Fourier integral \(F(\nu )\), equivalently called the finite continuous Fourier transform (CFT), is:
where the imaginary unity \(i=\sqrt{-1}\) is the base of complex numbers. This is a continuous/analog Fourier spectrum. Physically, it represents a distribution of the intensities as a function of linear continuous frequencies \(\nu \). Here, the term intensities refers to the intensities of the response function of the sample to the external perturbations. Integration is a smoothing operator. Thus, the fine details in c(t) are averaged over by integration in the CFT from (1).
In the Fourier signal processing, both t and \(\nu \) are equidistantly/uniformly discretized/digitized. Thus, with e.g. \(t=t_n=n\tau \, (0\le n\le N-1)\), a digitized FID is obtained as \(c(t)=c(n\tau )\equiv c_n\), where \(\tau =T/N\) is the sampling rate (or dwell time). Number N is the total length of the FID (or the total duration of the FID, or the total acquisition time for encoding a single time transient). The bandwidth BW and \(\tau \) for a complex-valued FID (as is the case in the quadrature encoding modality from MRS) are connected by the relation \(\textrm{BW}=1/\tau .\) The desired frequency content in \(c_n\) is determined by the chosen bandwidth.
A similar frequency discretization is \(\nu =\nu ^\mathrm{\scriptscriptstyle F}_k=k/T\, (0\le k\le N-1)\). This procedure discretizes the continuous response function \(F(\nu )\) as \(F(\nu )=F(k/T)\equiv F_k\), which is a stick/bar spectrum (i.e. it is defined only at the Fourier grid/mesh points \(\nu ^\mathrm{\scriptscriptstyle F}_k\)). With t and \(\nu \) discretized, the CFT, \(F(\nu )\), is mapped to the discrete Fourier transform (DFT), \(F_k\), which is simply the Riemann sum for (1):
where use is made of the relation \(N=T/\tau \). For \(k=1\), the Fourier grid \(\nu ^\mathrm{\scriptscriptstyle F}_1=1/T\) represents the Fourier frequency resolution. Thus, the larger values of T are necessary for an approximately adequate resolution in the Fourier spectrum \(F_k\). Still, even for a very long total acquisition time T, it is conceivable that none of the true resonant frequencies \(\{\nu _k\}\, (1\le k\le K)\) contained in each \(c_n\) would coincide with the Fourier grid frequencies \(\{\nu ^\mathrm{\scriptscriptstyle F}_k\}=\{k/T\}\, (0\le k\le N-1)\). Moreover, extended acquisition times T are impractical, particularly for FIDs encoded by MRS from patients.
The true (physical, genuine) frequencies \(\{\nu _k\}\, (1\le k\le K)\) characterize the given resonating system (atoms, molecules) in the scanned specimen of K constituents. That is why they are called the characteristic or eigen or fundamental frequencies. In signal processing, every such frequency is a complex quantity comprised of two real numbers, associated with the position and width of the resonance peak in the spectrum.
As it stands, the DFT is recognized as the trapezoidal numerical integration. This is the simplest numerical quadrature rule of the Fourier integral in (1). Complex quantity \(F_k\) is a single polynomial of degree \(N-1\) in the variable \(W_k\) with the expansion coefficients given by the FID data points \(\{c_n\}.\) Variable \(W_k\) is the undamped trigonometric oscillatory function, a sine and a cosine combined into the Euler formula for an exponential (an unattenuated complex harmonic), \(W^n_k=\textrm{e}^{-2\pi ink/N}=\cos {(2\pi nk/N)}-i\sin {(2\pi nk/N)}.\) The Fourier basis functions \(\{W_k\}\) satisfy their orthonormality relation:
where \(\delta _{k,k'}\) is the Kronecker \(\delta \)-symbol. This leads to the one-to-one correspondence \(F_k\leftrightarrow c_n\), which implies a complete equivalence between the time and frequency domain data in the Fourier signal processing. Such two representations are rooted in the two complementary or canonical conjugate variables, t and \(\nu \). Consequently, the same whole information is contained in \(\{c_n\}\, (0\le n\le N-1)\) and \(\{F_k\}\, (0\le k\le N-1)\). This feature is built into the inverse discrete Fourier transform (IDFT), which retrieves exactly the entire input data \(\{c_n\}\, (0\le n\le N-1)\), including noise:
The IDFT describes the time signal \(c_n\) as a linear combination of the undamped complex harmonics \(\{\textrm{e}^{2\pi ink/N}\}\) with complex amplitudes \(\{F_k\}.\) Since \(F_k\) is of a polynomial form for each k, it has no singularities. In particular, \(F_k\) is void of poles that yield the resonance peaks. These peaks are the signatures of spectra in MRS (as well as in other spectroscopies).
According to the time-frequency Heisenberg uncertainty principle \(\Delta t\Delta \nu \ge 1/(4\pi )\), good localization (compactness) is simultaneously unachievable in both the time and frequency domains. The most compact function in both the \(t-\) and \(\nu -\)domains is a Gaussian. For the precisely known intensity \(F_k\) at a fixed frequency k/T, there is no information whatsoever about any particular value of the time signal since the entire set \(\{c_n\}\, (0\le n\le N-1)\) is averaged over in (2). Likewise, at a given instant \(n\tau \), the time signal \(c_n\) is known, but it cannot be related to any specific intensity \(F_k\) because all of them \((0\le k\le N-1)\) are summed up in (4).
Despite the absence of the polar structure in the DFT, with a sufficient number of interferences of sines with cosines multiplying \(c_n\) of long length N, it may still become possible for \(F_k\) from (2) to roughly estimate at least some of the main spectral lineshapes using the given FID. The sine as well as the cosine functions are periodic and so are the harmonics \(\{\textrm{e}^{2\pi ink/N}\}\), implying that time signal \(c_n\) in the Fourier representation (4) is forced to be periodic. Thus, in the DFT, irrespective of the structure of the processed FIDs, all time signals are treated as if they were periodic. However, most measured time signals are not periodic.
In the DFT from (2), the product of the two sequences \(\{c_k\}\) and \(\{F_k\}\) of length N requires \(N^2\) multiplications. These become computationally time-consuming for large N. Advantageously, however, for a composite N, given e.g. by number 2 raised to a positive integer, the amount of multiplications can be enormously reduced. Stated more precisely, for \(N=2^\ell \, (\ell =1,2,3,\ldots )\), the mentioned sequence multiplications scale quasi-linearly with the FID length N as \(N\textrm{log}_2N\). This is the key feature of the Cooley-Tukey algorithm [69] for converting the DFT into the FFT, which revolutionized all fields that employ the Fourier-based signal processing, including analytical chemistry.
2.2 Usual derivative fast Fourier transform
The derivative discrete Fourier transform (dDFT) is obtained by applying the \(m\,\)th derivative operator \(\textrm{D}_m=(\text{ d }/\text{d }\nu )^m\) to the CFT from (1) and by subsequently discretizing t and \(\nu \). The result is represented by:
Derivative and integration operators have the opposite effects. Thus, by way of (5), the derivative operator \(\textrm{D}_m\) from (6) is anticipated to unsmooth the smoothing action of the CFT in (1). This would amount to achieving a higher resolution in the dDFT than in the DFT, as the applications to noiseless FIDs can indeed directly confirm.
The related derivative fast Fourier transform (dFFT) is the fast computation of the dDFT by the Cooley-Tukey algorithm [69], applied to the product of the time power function and the FID, i.e. \(\{(-2\pi it_n)^mc_n\}\, (0\le n\le N-1)\). The breakdown of the dDFT and dFFT for encoded FIDs is due to the multiplier \(t^m_n\) of \(c_n.\) The monomial \(t^m_n\) weighs most heavily the FID tail, which contains mainly noise in measured FIDs. Therefore, with the increased derivative order m, mostly noise is processed by the dFFT. This leads to information loss with a consequence of worsening both resolution and SNR [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67].
2.3 Optimized derivative fast Fourier transform
Solving the just stated problem, while preserving model independence and the Cooley-Tukey algorithm [69], would require an appropriate upgrade of the dDFT from (5). To that end, it is necessary to overcome the detrimental effect of the derivative-produced power function \(t^m_n=(n\tau )^m\). At larger \(n\tau \), the diverging behavior of \((n\tau )^m\) with the increased signal number n, at a fixed derivative order m, can be judiciously regularized. This is feasible through multiplication of \((n\tau )^m\) by an attenuating function of \(n\tau \). In an optimization procedure, the fastest decaying object functions multiplying \((n\tau )^m\) should preferably be used.
These functions are an exponential filter, the EF, \(\textrm{e}^{-\lambda _1(m,T)t_n}\) and a Gaussian filter, the GF, \(\textrm{e}^{-\lambda _2(m,T)t^2_n}\). Both filters decrease with the rising time t, i.e. they have the strictly positive damping parameters \(\lambda _p(m,T)>0\). They are ingrained in the adaptive power-exponential filter, the APEF:
and in the adaptive power-Gaussian filter, the APGF:
Employing either the discretized APEF or APGF would result in the optimized dDFT associated with the sequences \(E^{(m)}_k\) and \(G^{(m)}_k\):
respectively. Computations of the sets \(\{E^{(m)}_k\}\) and \(\{G^{(m)}_k\}\) by means of the Cooley-Tukey algorithm [69] yield two versions of the optimized dFFT, one with the APEF and the other with the APGF, respectively [68].
As has been demonstrated in Ref. [67] for MRS, the resulting combined effect of the power-exponential filter \((-2\pi it_n)^m\textrm{e}^{-\lambda _1(m,T)t_n}\) from (9) or the power-Gaussian filter \((-2\pi it_n)^m\textrm{e}^{-\lambda _2(m,T)t^2_n}\) from (10) in the optimized dFFT can lead to a hugely improved resolution and SNR relative to the FFT from (2) as well as compared with the usual dFFT from (5). For this to occur, the specific values of the damping parameter \(\lambda _p(m,T)>0\) for a fixed p are of practical importance.
The adverse consequences of the presence of the power function \(t^m_n\) are exacerbated for higher m. Therefore, the damping parameter \(\lambda _p(m,T)\) for a fixed p should be adapted to the increased m. In other words, a gradually enhanced damping should be employed to successively match the higher powers m of \(t_n\). Moreover, the damping parameter \(\lambda _p(m,T)\) should approximately be tailored to noise of the originally encoded FID so as to achieve simultaneously improved SNR and frequency resolution. Incorporating these aspects into the object or merit functions such as the EF, \(\textrm{e}^{-\lambda _1(m,T)t_n}\), or the GF, \(\textrm{e}^{-\lambda _2(m,T)t^2_n}\), can be made in a computerized optimization algorithm for a minimization procedure.
The Heisenberg uncertainty principle applies strictly to the FFT and yet resolution of this processor can be surpassed by another estimator. Resolution is higher in the optimized dFFT than in the FFT. In the optimized dFFT, the Heisenberg uncertainty principle appears in the form of the informational uncertainty principle: no richer information is possible to retrieve by reconstructions than that provided by the original FID [45, 46]. Resolution in the optimized dFFT is not predetermined by T, but rather by the spectral density (the number of the resonance peaks) in the analyzed chemical shift band. Moreover, the optimized dFFT also mitigates the resolution-SNR uncertainty relationship by means of the APEF or APGF, as explained.
2.4 Optimization of attenuation by analytical means
An alternative to the mentioned computerized optimization is a practical, quick and useful optimization by analytical means [67, 68]. It is based on a closed expression for \(\lambda _p(m,T)\) with a fixed p. This analytical formula is derived here after the salient preliminary considerations.
2.4.1 Resonance parameters in MRS
A crude estimate of the SNR in the encoded \(\{c_n\}\) is the ratio of the time signal intensities at e.g. \(t=0\) and \(t=T\), i.e. at the onset and at the end of the time-domain encodings, respectively. The decaying feature of a typical FID transient of total length \(N=T/\tau \) encoded by e.g. proton single-voxel MRS (from a sample containing a mixture of substances with various metabolites) is a linear combination of a number of attenuated complex exponentials of the type:
where \(\varphi _0\) is the phase offset at \(t=0\). Noise is implicitly assumed, but for brevity, is omitted in the explicit writing from (11). Here, \(T^\star _2\) is the exponential decay time constant:
where \(\gamma \) (in MHz/T) is the gyromagnetic ratio (quotient of the angular and magnetic moments).Footnote 1 Constant \(A>0\), as the magnitude of the initial intensity of the given FID, i.e. \(\left| c(t)\right| \) at \(t=0\), is directly proportional to the magnitude of the bulk magnetization vector \(\textbf{M}\) of the sample. Therefore, \(\left| c(0)\right| \) is also directly proportional to the number of the resonating protons (contributing to the time-domain response of the sample to the external excitation) and, hence, to the metabolite concentration.
Linear fundamental frequency \(\nu _0\) (in Hz) from (11) refers to the characteristic oscillations of the excited protons. Quantity \(T_2\) (in ms) from (12) is the natural transversal relaxation time from the spin-spin interactions of the resonating protons. In reality, \(T_2\) is perturbed by several factors, including the static magnetic field inhomogeneity \(\gamma \mathrm{\Delta } B_0\) (in Hz), as seen in (12). Generally, \(\mathrm{\Delta } B_0\) can be positive or negative. For \(\gamma \mathrm{\Delta } B_0>0\), there will be a line-breadth widening in the peak from the frequency spectrum associated with (11).
The perturbed value of \(T_2\) is called the effective transversal time and denoted by \(T^\star _2\) (in ms). This is the time needed to elapse before the bulk magnetization \(\textbf{M}\) of the excited sample attains the equilibrium distribution (alignment with the externally applied static polarizing magnetic field of strength \(B_0\)). The reciprocal \(1/(\pi T^\star _2)\) is the FWHM for an absorptive bell-shaped symmetric Lorentzian peak (\(\varphi _0=0\)) in the spectrum computed using (11):
Therefore, the FID from (11) can equivalently be written in terms of the FWHM as:
The same FWHM also enters the definition of the peak height and peak area of an absorptive Lorentzian profile, respectively:
2.4.2 A measure of the closeness of the FID decay to zero
For insufficiently long T (as is the case in FID encodings by MRS in the clinic), the time signals may have not decayed to zero at \(t=T\). This amounts to a truncation of the FID. Truncation of c(t) is represented as \(c(t)\textrm{H}(T-t)\), where \(\textrm{H}(T-t)\) is the Heaviside step function. This rectangular function is also called the rec-function or the box-function, which is equal to 1 and 0 for \(t\le T\) and \(t>T\), respectively. The Heaviside step function is the continuous counterpart of the Kronecker symbol from (3). The CFT from (1) with c(t) replaced by \(c(t)\textrm{H}(T-t)\) represents a convolution. A convolution in the time domain corresponds to a product in the frequency domain.
The Fourier transform of the rec-function is the sinc-function, which is defined as \([\sin {(\pi \nu )}]/(\pi \nu )\). Thus, a truncation of c(t) yields the product of the Fourier spectrum and the sinc-function. Since the sinc-function decays in an oscillatory manner, every peak in the CFT, \(F(\nu )\), computed from (1) with \(c(t)\textrm{H}(T-t)\), will be distorted, including its base and the wings/tails. This phenomenon is known as the Gibbs ringing. A potential reduction of such lineshape deformations would be important because they impede on extracting the reliable quantitative spectral information (e.g. peak areas and concentrations of metabolites in the tissue or biofluid scanned by MRS).
It is of practical interest for optimization to roughly quantify the closeness to zero of the FIDs at the end of encodings \((t=T)\). Such FIDs can be considered to have decayed sufficiently close to zero at \(t=T\) if therein the intensity of a typical transient c(T) from (11) is less than or equal to about 5% of the associated initial intensity at \(t=0\). From here, the condition that the decaying part \(A\textrm{e}^{-T/T^\star _2}\) of the FID in (11) drops to about 5% of A (i.e. to become 0.05A or A/20), would read as \(A\textrm{e}^{-T/T^\star _2}=0.05A\), which corresponds to \(T/T^\star _2\approx 2.9957\).
Simplifying then, by setting \(T/T^\star _2=3\), we have \(\textrm{e}^{-T/T^\star _2}=\textrm{e}^{-3}\approx 0.0498\), where rounding of 0.0498 to 0.05 approximately yields the sought 5% of the initial intensity A of FID (\(t=0\))Footnote 2. Instead of having SNR with the fixed percentage as \([A(0)/A(T)]\times 100\%=5\%\), or the fraction 0.05, we can take a more generic fraction \(\rho >1\) (as the selected threshold for considering that the FID tail is sufficiently immersed into noise) to rewrite the just stated condition as:
which then implies
For \(\rho >1\), it follows that \(\ln {\rho }>0\), which is compatible with the necessary physical positive-definiteness \((T/T^\star _2>0)\) of the lhs of Eq. (18). Number \(\rho \) is proportional to SNR of the input FID. In the encoded c(t) for \(0\le t\le T\), noise prevails when t tends to T. In other words, the FID closely matches the noise level near the end of the total acquisition time T. Thus, condition (17) means that the distortion of the FID due to the truncation artifacts is smaller than the noise level in the encoded FID at \(t=T\).
Quotient \(T/T^\star _2\) represents the extent of damping of c(t) from (11). An undamped transient in the FID corresponds to \(T/T^\star _2=0\). A nonzero finite damping, like the discussed attenuation rate \(T/T^\star _2=3\), refers to a transient which is reasonably well relaxed (i.e. embedded in noise) since it decayed to about 5% of its initial intensity at the onset of encodings \((t=0)\). An idealized, unrealistic FID with \(T/T^\star _2=\infty \) would be completely damped (fully relaxed), in the sense of exactly coinciding with zero at the end of encodings, i.e. \(c(T=\infty )=0\), as per (11).
2.4.3 Adaptive filters
The EF and GF themselves improve SNR and reduce the FID truncation artifacts. Thus, they belong to the category of denoising functions. Both filters perform smoothing of the given FID. Noise can, to a certain extent, be averaged out by smoothing. However, the EF and GF lead to resolution loss due to line broadening. The widened spectral profiles enhance the peak overlaps and this decreases resolution. Such an effect hampers quantification of the closely spaced resonances of interest. On the other hand, while the power function \(t^m\) greatly improves resolution for noise-free FIDs [55], this same monomial lowers SNR for encoded noise-corrupted FIDs and, by implication, deteriorates resolution. Hence, such two diametrically opposite trends from \(t^m\) and EF or GF ought to be reconciled for the success of an optimization.
The situation is salvaged by combining the two functions as their products through e.g. the APEF, \(t^m\textrm{e}^{-\lambda _1(m,T)t}\) or the APGF, \(t^m\textrm{e}^{-\lambda _2(m,T)t^2}\). In these products, the power function and an attenuated exponential (or a Gaussian) filter are designed to compensate their individual deficiencies. More precisely, noise produced by \(t^m\) should be damped by \(\textrm{e}^{-\lambda _p(m,T)t^p}\) in the combination \(t^m\textrm{e}^{-\lambda _p(m,T)t^p}\) for the chosen p.
Conversely, the line broadening effect caused by the attenuation function \(\textrm{e}^{-\lambda _p(m,T)t^p}\) is supposed to be balanced/mitigated by \(t^m\). Of course, such two-way compensations are limited by the SNR of the input FID. Since \(t^m\) itself amplifies noise with augmented m, a trade-off is needed for determining the practical upper limit of the derivative order to secure the acceptable spectral lineshapes amenable to a reliable quantification.
For this to occur with a model weighting function, it is natural to first implement the exponential filter, the EF, because an FID transient from encoding by MRS is itself exponentially damped, as per (11). Nevertheless, it is convenient to consider the EF and GF in concert by keeping p as a generic number (i.e. without fixing it in advance to the particular values \(p=1\) or \(p=2\)).
To proceed further, the following condition is of key importance for analytically optimized filters [68]:
where \(\alpha _p>0\) is a constant which is adapted to SNR of the original FID. The condition (19) at \(t=T\) acquires the form \((T\textrm{e}^{\alpha _p})^m=\textrm{e}^{\lambda _pT^p}\). Taking the natural logarithm of both sides of the latter expression, it follows \(m\ln {(T\textrm{e}^{\alpha _p})}=\lambda _pT^p\). This gives the formula for the sought damping parameter \(\lambda _p\equiv \lambda _p(m,T)\):
In particular, the constant \(\alpha _1\) in (19) plays the role of the amount of damping \(T/T^\star _2\) in c(t) from (11) at \(t=T\). Specifically, for \(m=1\), the cut-off factor \(\textrm{e}^{-\alpha _1}\) in (19) is reminiscent of \(\textrm{e}^{-T/T^\star _2}\) from (11) at \(t=T\). Further, observe that, similarly to the monomial \(t^m\), the corrective term \(\textrm{e}^{-\alpha _p}\) is also taken to the same power \(m\ge 1\) to become \(\textrm{e}^{-m\alpha _p}\) in (19). This, in turn, implies that the larger amount of noise produced by \(t^m\) in \(t^m\textrm{e}^{-\lambda _p(m,T)t^p}c(t)\) is countered by a stronger damping due to the filter \(\textrm{e}^{-\lambda _p(m,T)t^p}\) where \(\lambda _p(m,T)=(m/T^p)\ln {(T\textrm{e}^{\alpha _p})}\) as per (20).
Overall, it is the powered corrective term \(\textrm{e}^{-m\alpha _p}\) on the rhs Eq. (19) that adapts the ensuing filter \(t^m\textrm{e}^{-\lambda _p(m,T)t^p}\) to both the derivative order m and SNR of the FID. For a fixed p, this simultaneous adaptation is achieved by introducing a single positive parameter, which is \(\alpha _p\).
Therefore, for the two cases of particular interest (EF: \(p=1\) and GF: \(p=2\)) of the general filter \(\textrm{e}^{-\lambda _p(m,T)t^p}= \textrm{e}^{-m\mu _pt^p}\), the APEF and APGF from (7) and (8), can now acquire their digitized explicit forms:
where the relation \(\tau /T=1/N\) is used. Recall that integers n and m in (21) and (22) are the time signal numbers (\(n=0,1,\ldots ,N-1)\) and the derivative order (\(m=1,2,3,\ldots )\), respectively.
Notice that besides the generally unequal constants \(\alpha _1\) and \(\alpha _2\), the expressions for the APEF and APGF from (21) and (22) contain two other different terms \(n\tau /T=n/N\) and \((n\tau /T)^2=(n/N)^2\) that multiply \(m\ln {(T\textrm{e}^{\alpha _1})}\) and \(m\ln {(T\textrm{e}^{\alpha _2})}\), respectively.
In Ref. [68], we used the notations \(\{\lambda _\mathrm{\scriptscriptstyle E},\alpha \}\) (exponential damping) and \(\{\lambda _\mathrm{\scriptscriptstyle G},\beta \}\) (Gaussian damping) that now correspond to \(\{\lambda _1,\alpha _1\}\) and \(\{\lambda _2,\alpha _2\}\), respectively. The notations \(\{\lambda _1,\alpha _1\}\) and \(\{\lambda _2,\alpha _2\}\) permit a more compact writing since the single expression \((-2\pi in\tau )^m\textrm{e}^{-m(n\tau /T)^p\ln {(T\textrm{e}^{\alpha _p})}}\) can be either the APEF \((p=1)\) or the APGF \((p=2)\). Moreover, p need not be restricted to 1 or 2 alone. Of course, for any p, the associated \(\alpha _p\) must always be a positive number, \(\alpha _p>0\), as stated with (19).
2.4.4 The width of the adaptive power-exponential filter
It is deemed illustrative to specify some parameters from the preceding exposition with their quantitative values regarding e.g. the line broadening (LB) and the corresponding time constant (TC) [28]. The aim is to see whether a case of the matched filter could emerge, signifying that the TC of the APEF is close to the spin-spin relaxation time \(T^\star _2\) of a metabolite physically present in the scanned tissue or biofluid.
To that end, consider an example with the APEF (\(p=1\)), which uses the EF \((\mu _p=\mu _1,\, \alpha _p=\alpha _1\)) in the attenuation function \(\textrm{e}^{-m\mu _1n\tau }\). An attenuation of the FID through multiplication of c(t) from (11) by \(\textrm{e}^{-\lambda _1(m,T)t}\) gives:
where the line broadening \(\textrm{LB}\) (in Hz) represents the width of the APEF,
The corresponding time constant \(\textrm{TC}\) (in ms) of the APEF is the reciprocal of \(\pi \textrm{LB}\) and thus equal to \(1/\lambda _1(m,T)\):
Comparing (12) and (24), it follows that the filter \(\textrm{e}^{-\lambda _1(m,T)t}\) acts as yet another perturber, which further broadens the natural relaxation time \(T_2\) on top of the already existing line-breadth widening due to the magnetic field inhomogeneity for \(\mathrm{\Delta }B_0>0\):
To exemplify the LB and TC factors, let us consider the acquisition parameters of the FIDs encoded at 1.5T clinical scanner from white matter in the brain of a 25 year old healthy male volunteer (as in Sect. 3). These are short FIDs (total length \(N=512\)), sampled at rate \(\tau =1\,\textrm{ms}\) (BW=1000 Hz) for which \(T=N\tau =512\,\textrm{ms}\). To obtain the damping time constant \(\textrm{TC}\) in (26) and the line broadening factor \(\textrm{LB}\) in (25) for the EF part \(\textrm{e}^{-m\mu _1n\tau }\) of the APEF, we choose \(\alpha _1=3\), as in the already discussed example \(T/T^\star _2=3\) associated with (11), and set \(m=1\) (the first derivative).
The \(\textrm{LB}\) and \(\textrm{TC}\) parameters of this specific APEF are calculated from (25) and (26) with the results \(\textrm{LB}=[1/(\pi T)]\ln {(T\textrm{e}^{\alpha _1})}=1.4489\,\textrm{Hz}\) and \(\textrm{TC}=1/(\pi \textrm{LB})=219.6888\, \textrm{ms}\), respectively. The latter finding of about 220 ms for \(\textrm{TC}\) (corresponding to the \(\textrm{LB}\) of about 1.45 Hz) is close to the effective relaxation time \(T^\star _2\approx 217\,\textrm{ms}\) of metabolite creatine (Cr), \(\mathrm{N(CH_3)}\), resonating at 3.03 ppm (parts per million) [28].
For \(m\ge 2\), the values of \(\textrm{LB}\) and \(\textrm{TC}\) will rise and fall, respectively. Thus, for e.g. \(m=2\), we have \(\textrm{LB}=2.8978\) Hz (\(\textrm{TC}=109.8444\) ms). On the other hand, for \(m=4\), it follows \(\textrm{LB}=5.7956\) Hz (\(\textrm{TC}=54.9222\) ms). By referring to free choline (Cho), \(\mathrm{N(CH_3)_3}\) and water, \(\mathrm{H_2O}\), resonating at 3.19 ppm and 4.70 ppm (temperature \(37^\circ \)), the values of \(T^\star _2\) are 162 ms and 60-130 ms, respectively [28]. Thus, as the derivative order m is augmented, the damping time constant \(\textrm{TC}\) of the exemplified APEF is decreasing and attaining its values that range between the relaxation times \(T^\star _2\) of Cr and \(\mathrm{H_2O}\).
The usual matched filters (either an exponential or a Gaussian) for a chosen damping parameter can describe only one metabolite (including, eventually, its very small neighborhood). It would then be tempting to think that for a fixed m in the optimized dFFT, a single datum for the TC parameter from the APEF could likewise match the \(T^\star _2\) value of one metabolite alone (and possibly a very few adjacent resonances assigned to different metabolites). However, practice shows that the optimized dFFT (at a single m yielding the stabilized lineshape) using the APEF or APGF can accurately describe many resonance profiles from the entire chemical shift band of the main interest, e.g. 0.0-4.25 ppm (see Refs. [67, 68] and the present Sect. 3).
The currently elaborated general forms of the damping parameters \(\lambda _1(m,T)\) and \(\lambda _2(m,T)\) in APEF and APGF, respectively, from the optimized dFFT have recently been used in Ref. [67]. Therein, the optimized dFFT for the FIDs encoded with and without water suppression by in vitro proton MRS at a 1.5T clinical scanner resulted in a powerful performance yielding simultaneously improved resolution and SNR relative to the FFT. Similarly successful applications of the optimized dFFT, with narrowed lineshapes and enhanced heights of physical resonances, have also been reported [67] by employing the APEF for an FID encoded with water suppression by in vitro proton MRS at a 600 MHz (\(\approx \)14.1T) Bruker spectrometer.
2.4.5 Normalized magnitude spectra
Given that the peak heights keep on rising with augmented m, it is impractical to monitor stabilization in the derivative lineshape developments for the successive derivative orders m. This obstacle is easily surmounted by using the normalizing magnitude lineshapes. A normalized spectrum \(\left| \textrm{D}_{m}\textrm{FFT}\right| _\textrm{N}\) \((m>0)\) is obtained by scaling the derivative spectrum \(\left| \textrm{D}_{m}\textrm{FFT}\right| \, (m=1,2,\ldots )\). Firstly, in a narrow chemical shift band around a selected peak, for a fixed derivative order m, we extract the maximum value \(\textrm{max}\left| \textrm{D}_{m}\textrm{FFT}\right| \) of the sequence \(\left| \textrm{D}_{m}\textrm{FFT}\right| \) from its set at the N Fourier grid frequencies \(\nu ^\mathrm{\scriptscriptstyle F}_k\, (0\le k\le N-1)\):
This relation includes the nonderivative case (\(m=0\)) as \(\left| \textrm{FFT}\right| ^\textrm{max}\), which itself is not normalized. Secondly, the scaling factors \(\textrm{R}_{m}>0\) are defined as:
Thirdly, the normalized spectrum is set up by the relation:
This, on account of (29), can equivalently be rewritten according to:
Therefore, for each fixed positive m, the normalized magnitude derivative spectrum \(\left| \textrm{D}_{m}\textrm{FFT}\right| _\textrm{N}\) is the product of the unnormalized spectrum \(\left| \textrm{D}_{m}\textrm{FFT}\right| \) with \(\left| \textrm{FFT}\right| ^\textrm{max}/\left| \textrm{D}_{m}\textrm{FFT}\right| ^\textrm{max}.\) The latter quantity is the ratio of the maximal nonderivative (\(m=0\)) and the maximal unnormalized derivative spectra \((m>0\)). The notations (28)-(31) refer explicitly to the optimized dFFT with either the APEF \((p=1)\) or the APGF \((p=2)\). The same relation can, of course, be also employed for the unattenuated dFFT which, however, will not be shown in Sect. 3 because of its inferior performance [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68].
3 Results and discussion
Standard single-voxel in vivo proton MRS was employed to encode the FIDs from white matter in the brain of a 25 year old healthy male volunteer. Encodings with and without water suppression have been made at the Karolinska Hospital in Stockholm, using a 1.5T General Electric (GE) clinical scanner with the conventional point-resolved spectroscopy sequence (PRESS). The acquisition parameters consist of the Larmor frequency \(\nu _\textrm{L}=63.8646\,\textrm{MHz}\) for the static magnetic field strength \(B_0=1.5\textrm{T}\), the bandwidth \(\mathrm{BW=1000\, Hz}\), the FID full length \(N=512\), the sampling rate or the dwell time \(\tau =1/\mathrm{BW=1\,ms}\), the repetition time \(\mathrm{TR=2000\, ms}\), the number of excitations \(\mathrm{NEX=128}\) and the echo times \(\mathrm{TE=13, \,23}\) and \(\mathrm{272\, ms}\).

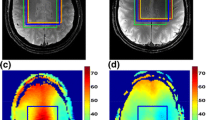
In vivo MRS for white matter in the brain of a 25 year old healthy male volunteer. Multiple time signals or FIDs have been encoded with and without water suppression by single-voxel in vivo proton MRS at a GE clinical scanner (1.5T). The acquisition parameters were: \(N=512\), NEX = 128, BW = 1000 Hz, \(\tau =1\, \textrm{ms}\), TR = 2000 ms and TE = 272 ms. The encoded raw 128 FIDs were averaged. The averaged FIDs (a, b; e, f) are not zero-filled, nor modified in any other way (no multiplying weight function, no phasing, no eddy current corrections, etc.). The FFT magnitude spectra (c, d) and (g, h) are for the averaged FIDs that are, however, extrapolated to 2N by one zero filling. The spectral intensities on the ordinates are in arbitrary units (au). Resonance frequencies (chemical shifts) on the abscissae are in dimensionless units, parts per million (ppm). For details, see the text
As is customary, the applied quadrature encoding provided the two-channel FIDs, one for the real (Re) and the other for the imaginary (Im) parts. The usual time signal arithmetic averaging for each set of the acquired 128 FIDs is made in the scanner to improve the signal to noise ratio, SNR. It is these averaged FIDs that are subjected to our data analysis. Among the mentioned three echo times, the present illustrations are focused upon \(\mathrm{TE=272\, ms}\) alone (from a different aspect, the results for \(\mathrm{TE=13}\) and 23 ms will be reported separately).
Figure 1 shows the encoded time signals alongside the corresponding reconstructed FFT spectra in the usual nonderivative form (\(m=0\)). The displayed FIDs represent the raw data of the original length (512 ms) with no correction whatsoever. However, prior to passing to the frequency domain, these FIDs are zero-filled once to compute the shown FFT spectra. In Fig. 1, the left and right columns are for the FIDs encoded without and with water suppression, respectively. The top two pairs of panels (a, b; e, f) are for the FIDs, whereas the associated couple of the bottom panels (c, d; g, h) are for the FFT spectra in the magnitude mode (no weighting function multiplying the processed FIDs). The real parts of the complex-valued FIDs are on panels (a, e), while panels (b, f) are for the imaginary parts of the time signals. For convenience, the ordinates on panels (e, f) are multiplied by a factor of 100.
It is seen that the lines on panels (a, b) for the water-unsuppressed FIDs represent mainly the smooth time-domain envelopes. In these waveforms, there is only a broad-shaped dip on panel (a), whereas on panel (b), merely a saturation-type waveform is seen for the increased time. The reason for such structureless patterns is in the usual dominance of the water concentration with no hint about the actual presence of any other constituents. The water response to the external excitations during the MRS scans is the strongest among all other metabolites because of the dominance of the \(\mathrm{H_2O}\) molecules in the brain tissue.
On the other hand, the water-suppressed FID waveforms (e, f) are structured, showing very sharp oscillations superimposed on the time-domain envelopes. With the water abundance significantly reduced (by the standard inversion recovery procedure) during the measurements, such structures clearly emerge. This could arguably open the chance to peer more deeply into the fuller content of the FIDs acquired with water suppression. Of course, the sought content of the water-suppressed FIDs is still opaque and inaccessible to a direct interpretation by inspecting solely the drawings of the time domain data on panels (e, f).
Looking at panels (a, b) or (e, f), it follows that at any given time point, say \(t=n\tau \), on the abscissae, the FID intensities \(\{\textrm{Re}(c_n),\, \textrm{Im}(c_n)\}\) on the ordinates are well defined (minimal uncertainties), respectively. On the other hand, there are maximal uncertainties in spectra \(\left| F_k\right| \) on panels (c, d) or (g, h) (and indeed from the entire Nyquist range) as to which frequencies have actually generated the specific values \(\{\textrm{Re}(c_n),\, \textrm{Im}(c_n)\}\), respectively. This is a consequence of the time-frequency Heisenberg uncertainty principle since, as stated in Sect. 2, all the N frequencies k/T from Eq. (2) contributed to any given value of \(c_n\).
The usefulness of the information-preserving frequency-domain representation of the encoded time signals, as shown by the FFT spectra (c, d; g, h), offers an exploratory avenue for data analysis, which complements the message conveyed by panels (a, b; e, f). This approach with the FFT is more amenable to at least a preliminary interpretation. Panels (c, g) are for a wider chemical shift band 1.75-6.0 ppm, which includes the water peak located at 4.68 ppm. Thus, on panel (c) for the water-unsuppressed FID, besides the giant and perfectly symmetric water singlet resonance peak with its long tail nothing else is noticeable.
The situation changes markedly for the FFT spectrum on panel (g) with the water-suppressed FID, where some additional small peaks appear. These peaks are assigned to the molecules of nitrogen-acetyl aspartate (NAA), total creatine (tCr) and total choline (tCho) that resonate at their characteristic frequencies near 2.0, 3.0 and 3.2 ppm, respectively. The tCr metabolite is comprised of creatine and phosphocreatine, Cr and PCr, respectively. The tCho metabolite contains the free choline, phosphocholine and glycerophosphocholine, Cho, PC and GPC, respectively.
There is also a more spread-out spectral structure near 3.95 ppm, a part of which is assigned to yet another tCr. This small portion of the spectrum is most heavily distorted because it is the closest neighbor of the still large water residual peak at 4.68 ppm. Especially on the base (bottom, foot), the residual water peak on panel (g) does not preserve the ideal symmetry of its predecessor, which is the \(\mathrm{H_2O}\) resonance on panel (c).
Panels (d, h) for the FFT spectra are for a smaller chemical shift sub-band 1.75-4.25 ppm, which excludes the water peak. Even in this zoomed frequency window, the spectrum on panel (d) for the water-unsuppressed FID persists to be uninformative in its entirety. It shows only some glimpses of the severely deformed minuscule spectral humps close to the locations of NAA (\(\sim \)2.0 ppm), tCr (\(\sim \)3.0 ppm) and tCho (\(\sim \)3.2 ppm). These lineshape profiles are glued to a considerably elevated background, which stems mostly from the long-extended tail of the water resonance and partly from macromolecules (proteins,...).
On panel (h) for the water-suppressed time signal, the FFT envelope takes a form which is more reminiscent of the normal-appearing total shape spectrum for the white matter of the brain in a healthy adult. Besides a clear visibility of the peaks for NAA (\(\sim \)2.0 ppm), tCr (\(\sim \)3.0 ppm) and tCho (\(\sim \)3.2 ppm), there is also a small supplement set of the resonance-type spectral structures. They are positioned at 2.3-2.6 ppm and assigned to the multiplets of NAA and Glx, where the latter notation refers to the sum of the contributions from glutamine (Gln) and glutamate (Glu). Moreover, the multiplet of myo-inositol (m-Ins) can be noted at 3.5-3.7 ppm. This is followed by the widened structure at 3.9-4.25 ppm to which the other mentioned tCr resonance is amalgamated near 3.95 ppm. The irregularly clustered step-wise shape of the wide spectral structure at 3.9-4.25 ppm indicates a significant extent of distortion produced by the incomplete water suppression in the process of the FID encodings.
All these spectral profiles on panel (h) are asymmetric and strongly deformed, particularly at their bottom sections. There are several reasons for such an occurrence. They include the presence of the said large residual water peak as well as broader macromolecules, constructive/destructive interference of the real and imaginary parts of the FFT magnitude spectrum, tightly overlapped lineshapes of the adjacent resonances due to similar spin-spin relaxation times of many tissue metabolites, etc. These lineshape distortions are further exacerbated by the ever present phase mismatch of the neighboring resonances. The phase disparities among various resonances are attributed to different lineshape modifications caused by a time delay between the end of the excitation pulse and the beginning of the FID encodings.
No spectral mode, including the magnitude mode, is immune to such phase mismatching. To roughly rectify the deformed lineshapes of the real quasi-absorptive part of a complex spectrum, the encoded FIDs are customarily multiplied by a zero-order frequency-independent phase. However, no single phase correction can simultaneously ameliorate the unequally phase-distorted lineshapes of all the peaks located at different resonance frequencies. Moreover, an FID phasing can lead to resonance inversion, i.e. to converting some of the upward-oriented peaks to the peaks pointing downward. Further, the zero-order phase correction even with a minimal background baseline does not yield the strictly positive-definite envelope throughout the frequency range of interest. By definition, the magnitude mode is positive-definite and hence insensitive to any zero-order phase correction of either the complex spectrum or the complex time signal.
Overall, Fig. 1 shows that the FFT can yield only crude metabolic information. For the FIDs encoded without water suppression, the FFT is meaningful only for the water molecules. On panel (c), the spectroscopic data from the water resonance could be approximately parametrized by extracting the FWHM, peak height and peak area. This would be a rough estimate as it does not rule out the realistic possibility that even the isolated and presumed structureless bell-shaped peaks could hide some other resonances [64, 65]. In order to use water as an internal reference metabolite, the computed dimensionless area of the \(\mathrm{H_2O}\) peak is to be calibrated, i.e. expressed in the appropriate biochemical units (e.g moles per kilogram, M/kg or mole per liter, M/L).
This can be done through multiplication of the reconstructed water peak area by an independently known physical constant in biochemical units (e.g. M/kg or M/L) for the normal brain of the specified tissue and region. Such a calibration can establish water as an internal reference standard for concentrations. Subsequently, computing the other peak areas in the given spectrum would give the concentrations of the remaining assigned metabolites, quantitatively expressed also in the biochemical units (M/kg, M/L) as the percentage of the water concentration.
Evidently, the obtained results for all the other metabolites represent their relative concentrations because they are referred to another metabolite (e.g. water, as the internal standard, in this discussion). The same statement also applies to external standards, employed for samples from in vitro MRS as in e.g. Refs. [67, 68, 71] and elsewhere in the MRS literature.
However, for metabolites other than the \(\mathrm{H_2O}\) molecules, the FFT envelopes on panels (c, d) of Fig. 1 cannot be quantified even approximately by using the water-unsuppressed FID. This is the main reason for which the great majority of FID encodings are preferentially carried out with water suppression. Such an alternative leads to more informative FFT spectra, as is clear on panels (g, h). Nevertheless, despite this visual qualitative improvement relative to panels (c, d), no quantification is hardly possible on panels (g, h) either. The reason is in the considerable lineshape distortions and peak overlaps. For example, the peak area determinations by any quadrature rule cannot be done on panel (h) since no profile therein is amenable to its reliable enclosing/boxing within the well-defined chemical shift bounds for a numerical integration. It then follows that also for the FIDs encoded with water suppression, the FFT is not of much practical use for in vivo MRS.
Such a situation calls for some other types of signal processing in an attempt to go beyond the FFT (unweighted or weighted alike). To that end, while still staying within the Fourier-based estimations, the optimized dFFT is one of the promising candidates also for in vivo MRS. Such an expectation is based on the previous applications of this processor to in vitro MRS. The optimized dFFT, which starts as a shape estimator and finishes as a parameter estimator, has been shown to have an unprecedented performance for the FIDs encoded by in vitro proton MRS at both low and high magnetic fields [67] (1.5T) and [68] (14.1T).
Of particular relevance to the current study, this processor gave in Ref. [67] the comparable high-resolution spectra for the short FIDs (0.5KB) encoded from a Philips phantom [70] of excellent SNR by in vitro MRS with and without water suppression at a GE clinical scanner (1.5T). It is important to find out whether a similar success can be replicated for the present short noisy FIDs (0.5KB) encoded by in vivo MRS from a human brain also at a GE clinical scanner (1.5T) with and without water suppression. This would be significant, especially given the potential clinical ramifications mentioned in Ref. [67].
Thus, the remaining four figures are on derivative estimations by the optimized dFFT with \(1\le m\le 3\) using the FIDs encoded with and without water suppression. The FFT spectra (d, h) in Fig. 1 will be shown again as the reference/seed envelopes. To highlight the main metabolites of interest to diagnostics by MRS, all the spectra shall be depicted at frequencies below 4.25 ppm, i.e. without the presence of the water peak. The retrieved spectra for the two dramatically different FIDs from panels (a, b; e, f) of Fig. 1 will be compared with each other. In the computations, we employ both the adaptive power-exponential filter, the APEF, and the adaptive power-Gaussian filter, the APGF, with the varying damping/attenuation parameters \(\alpha _1\) and \(\alpha _2\) from Eqs. (21) and (22), respectively.
The derivative spectra for the FIDs with and without water suppression are jointly displayed in Figs. 2 and 3. They both refer to the first three derivative orders \((1\le m\le 3)\). This is followed by Figs. 4 (\(m=3\)) and 5 (\(1\le m\le 3\)) with five and two damping parameters, respectively. The APGF is on Figs. 2 (\(\alpha _2=5\)) and 4 (\(\alpha _2=1.75, \, 2.0, \, 2.25, \, 2.5, \, 5.0)\), whereas the APEF is on Figs. 3 (\(\alpha _1=3\)) and 5 (\(\alpha _1=1.5,\, 3\)). The purpose of this specific protocol is to test the sensitivity/stability of the optimized dFFT to the derivative order m and to the amount of damping \(\alpha _p\) in the APEF (\(p=1\)) and APGF (\(p=2\)). Thus, Figs. 2 and 3 vary the derivative order (\(m=1-3\)) for the fixed damping coefficient \(\alpha _2=5\) (Gaussian) and \(\alpha _1=3\) (exponential), respectively. On the other hand, Fig. 4 is for a fixed derivative order (\(m=3\)), while varying the Gaussian damping \((\alpha _2=1.75-5.0)\). Finally, Fig. 5 varies both the derivative order (\(1\le m\le 3\)) and the exponential damping (\(\alpha _1=1.5,\, 3.0\)).
To recall, we reemphasize that the amount of an exponential and a Gaussian damping \(\alpha _p\) in the APEF (\(p=1\)) and APGF (\(p=2\)), respectively, represents an approximate measure of SNR through the closeness of the decline of the encoded FID to zero at the end of the total acquisition time, i.e. at \(t=T\). This factor \(\alpha _p\) enters the overall smoothing parameter \(\lambda _p\) in the given attenuation filter \(\textrm{exp}\{-\lambda _p(m,T)(n\tau )^p\}\) with \(\lambda _p(m,T)=(m/T^p)\textrm{ln}(T\textrm{e}^{\alpha _p})\).
Moreover, the smoothing parameter \(\lambda _p\) also controls resolution since it is proportional to the line broadening. Thus, the same parameter \(\lambda _p\) can judiciously balance SNR and resolution. To that end, a practical trade-off between SNR and resolution is necessary. According to Refs. [67, 68], it is possible to simultaneously improve SNR and resolution by reliance upon the analytically derived optimization (20) of \(\lambda _p\). In Eq. (25), the line broadening for the APEF (\(p=1\)) is denoted by \(\textrm{LB}= (1/\pi )\lambda _1(m,T)\).
The way this design works is fully explained in Sect. 2 and, to better follow the further exposition, it shall now be succinctly recapitulated. An attenuation filter broadens each spectral line by the same constant. This constant may be different for the EF and GF. As stated, both the EF and GF can improve SNR, but they reduce resolution. It should be remarked that even if the damping factors \(\alpha _1\) in the APEF and \(\alpha _2\) in the APGF are numerically identical, these two filters will have different smoothing parameters \(\lambda _1\) and \(\lambda _2\) on account of the relation \(\lambda _2(m,T)=(1/T)\lambda _1(m,T)\) valid for \(\alpha _1=\alpha _2\).
On the other hand, the purpose of derivative estimation is to increase both SNR and resolution in concert. Thus, for a better resolution, it seems counter-intuitive that the encoded FID should be weighted by a filter which, in turn, produces a line-broadening and enhances the resonance overlaps. However, the EF and GF, if implemented by the APEF and APGF, respectively, actually constitute a virtue of the optimized dFFT. In this processor, the following twofold feature operates synergistically: for noisy FIDs, the reduced SNR due to \(t^m\) is compensated by \(\textrm{exp}\{-\lambda _p(m,T)(n\tau )^p\}\) and the lowered frequency resolution due to \(\textrm{exp}\{-\lambda _p(m,T)(n\tau )^p\}\) is countered by \(t^m\).
As a net result, the optimized dFFT improves simultaneously SNR and resolution. Such an outcome is due to the usage of the product \(t^m\textrm{exp}\{-\lambda _p(m,T)(n\tau )^p\}\). This product minimizes the deficiencies of \(t^m\) and \(\textrm{exp}\{-\lambda _p(m,T)(n\tau )^p\}\), while maximizing the benefits furnished by these latter two separate weighting functions.

Single-voxel in vivo proton MRS for white matter in the brain of a 25 year old healthy male volunteer. For spectra, the averaged values of the encoded 128 FIDs are used. Nonderivative \((m=0)\) and derivative \((m>0)\) magnitude spectra for the water-unsuppressed and water-suppressed zero-filled FIDs are on panels (a-d) and (e-h) in the left and right columns, respectively. The FFT spectra (a, e) with \(m=0\) are with no filtering. The optimized dFFT spectra (b-d; f-h) with the derivative orders \(1\le m\le 3\) are normalized and refer to the adaptive power-Gaussian filter, the APGF, for the damping parameter \(\alpha _2=5\). The spectral intensities on the ordinates are in arbitrary units (au). Resonance frequencies (chemical shifts) on the abscissae are in dimensionless units, parts per million (ppm). For details, see the text
With the outlined refreshment, borrowed from Sect. 2, we can now pass onto Figs. 2-5. As announced, the first comparisons of the spectra from the optimized dFFT with the three derivative orders (\(1\le m\le 3\)) for the FIDs with and without water suppression are on Fig. 2 for the APGF at the Gaussian damping \(\alpha _2=5.0\). Such a strong FID attenuation with the ensuing enhanced lineshape smoothing in the derivative spectra is anticipated not to split apart the tightly overlapped resonances. This is indeed observed in Fig. 2, where the derivative spectra at \(1\le m\le 3\) in the optimized dFFT on panels (b-d) and (f-h) for the FIDs without and with water suppression, respectively, are relatively sparse.
The said spectral sparsity is partially due to the long echo time \(\textrm{TE}=272\,\textrm{ms}\) at which the short-lived resonances have either decayed or significantly reduced their intensities. However, the most important information is on the panels (a-d) from the left column of Fig. 2 displaying the spectra from the FID encoded without water suppression. The associated FFT spectrum (a) is impoverished as it barely visualizes only the three minuscule bumps for NAA, tCr and tCho riding on the elevated tail of the water peak.
This is contrasted to the corresponding derivative spectra for \(1\le m\le 3\) (b-d). The improvement is obvious already with the first derivative with \(m=1\) (b), where the smoothly decaying lineshape with \(m=0\) (a) is now segmented into a number of spectral structures. Therein, the most prominently occurring is the large NAA peak near 2.0 ppm. Further, one can identify the inverted two peaks of tCr (\(\sim \)3.0, \(\sim \)3.95 ppm). Likewise, the peaks of m-Ins (\(\sim \)3.6 ppm) and tCho (\(\sim \)3.2 ppm) are pointing downward. Also visible within 2.1-2.6 ppm is the smaller NAA peak as well as the two adjacent Glx resonances preceded by the peak of gamma amino butyric acid (GABA). All these weak-intensity structures ride on the water tail, which appears to be considerably lowered relative to panel (a).
The second derivative with \(m=2\) (c) visibly suppresses the ridge from the water tail around 4.0 ppm. As a consequence, below 3.75 ppm, the whole background baseline is brought down closer to the chemical shift axis. Above 3.75 ppm, the still elevated baseline leads to an overestimation of the tCr peak height near 3.95 ppm. All the earlier mentioned peaks on panel (b) remain on panel (c), but now they point upward. The peaks of NAA (\(\sim \)2.0 ppm), tCr (\(\sim \)3.0 ppm) and tCho (\(\sim \)3.2 ppm) are intense and well delineated. Below 2.0 ppm, there is no appreciable change in the low-lying spectral wiggles immersed in noise.
The 3rd derivative with \(m=3\) (d) markedly reduces the water tail, including its remaining ridge above 4.0 ppm. Such an effect further flattens the background baseline throughout the shown frequency range. This spectrum begins to exhibit the embryo of an additional Glx peak positioned near 2.15 ppm, climbing as a shoulder of the large NAA peak (\(\sim 2.0\) ppm). Note that the dip near 3.1 ppm between the tCr (\(\sim \)3.0 ppm) and tCho (\(\sim \)3.2 ppm) peaks has now fully descended to the chemical shift axis. This is a notable improvement relative to the corresponding elevated valley on panel (c).
Taken together, the overall performance of the optimized dFFT on panels (b-d) is commendable, in spite of the seemingly insurmountable challenges posed by the encoded water-unsuppressed FID. This is all the more remarkable, especially given the corresponding completely uninterpretable FFT spectrum with \(m=0\) (a). The first derivative with \(m=1\) (b) sets the stage towards the further improvement. Subsequently, the steady progress is secured by the second and third derivatives with \(m=2\) (c) and \(m=3\) (d), respectively.
As to the right column of Fig. 2, the optimized dFFT with \(1\le m\le 3\) (f-h) is tasked to refine the FFT (e). This is easier because of the usage of the water-suppressed FID for which the FFT (e) itself is not unreasonable. Already the first derivative with \(m=1\) (f) delivers an adequate lineshape, which brings the background very close to the chemical shift axis at all the displayed frequencies. The improvement on panel (f) relative to panel (e) is observed everywhere and most notably at the end of the range, in the vicinity of 4.0 ppm. Therein, with just its first derivative (\(m=1\)), the optimized dFFT (f) maps the irregular and highly deformed spectral structure around 3.95 ppm from panel (e) into a well-delineated tCr peak.
Moreover, unlike some of the inverted and tilted peaks on panel (b), superimposed on the water tail, which keeps on rising with the augmented frequencies, all the peaks on panel (f) are pointed upward and lying straight on the flat background. This is the result of a more marked narrowing of the bottom/base of the \(\mathrm{H_2O}\) peak (not shown) in the first derivative spectra from the water-suppressed than for the water-unsuppressed FIDs.
From the jagged left hand side of the broad spectral structure around 4.0 ppm on panel (e), there is only a small leftover on panel (f). However, this remainder, which distorts the lower left part of the tCr peak at 3.95 ppm, is strongly diminished by the second derivative with \(m=2\) (g). Comparing the left and right columns, it is observed that for most of the shown frequencies (i.e. within 0.0-3.75 ppm), the second derivative spectra on panels (c) and (g) without and with water suppression in the FIDs, respectively, are quite alike. The significant difference between these two spectra occurs only in a small range above 3.9 up to 4.25 ppm. This remaining discrepancy is due to the distorting effect of the unsuppressed water in the FID processed on panel (c).
By going to the third derivative with \(m=3\) (h), the resulting lineshape is seen to be very similar to the predecessor spectrum with \(m=2\) (g). This points to a relative stability with no derivative-induced spectral distortions for the increased derivative order m. In fact, all the presently considered derivative orders are low \((m\le 3)\). Near 2.15 ppm, the slight left shoulder of the NAA peak from panel (g) begins to form another Glx resonance. Importantly, throughout the analyzed frequency interval, there is also a very good agreement between the two derivative spectra with \(m=3\) on panels (d) and (h) for the FIDs without and with water suppression, respectively.
Such agreement is on the level of the shape of the spectra on panels (d, h). The dimensionless ordinates in arbitrary units show the values of the spectral intensities that are numerically different in the optimized dFFT for the FIDs without (d) and with (h) water suppression. This is not unexpected. Firstly, the intensities in the corresponding seed spectra from the FFT (to which the derivative operator \(\textrm{D}_m\) is applied) on panels (d, h) of Fig. 1 differ even much more appreciably. Secondly, the numbers in arbitrary units on the ordinates carry no physical information, which is obtainable only after the appropriate calibration, as mentioned.
Moreover, since the ordinates are dimensionless, the approximately converged spectra in the optimized dFFT on panels (d, h) of Fig. 2 might as well be normalized to each other. In such a case, most of the individual peak areas on these panels for the FIDs without and with water suppression would turn out to be very similar to each other. So would be the related metabolite concentrations in biochemical units. This is what matters diagnostically in MRS, and not some numbers in arbitrary units on the ordinates. In the MRS literature, the ordinates in spectra are most frequently drawn with no numbers at all.
For in vivo MRS, agreement between panels (d, h) of Fig. 2 cannot be understated. It corroborates the like conclusion regarding in vitro MRS from Ref. [67]. The latter study was on the optimized dFFT for the FIDs encoded with and without water suppression from a Philips MRS phantom [70]. Still, the stated successful outcome from comparing panels (d, h) of Fig. 2 is not the end-point of the analysis as there is still room for further improvements. Namely, the intense Gaussian damping \(\lambda _2=5\) on panels (d, h) effectively precludes achieving a finer resolution with more spectral details.
In Fig. 3, we switch to the other filter, i.e. the APEF and choose the exponential damping \(\alpha _1=3\). In this setting too, we apply the optimized dFFT to the same FIDs encoded with and without water suppression. Herein, however, a shorter chemical shift band is considered, which unlike Fig. 2, now starts from 1.75 ppm. The reason is in Fig. 2, where the sub-band 0.0-1.75 ppm shows that the lineshapes on both the left and right columns are embedded in the background with no pronounced resonances to be assigned to the known metabolites. For the same reason, Figs 4 and 5 will begin with 1.75 ppm, as well.

Single-voxel in vivo proton MRS for white matter in the brain of a 25 year old healthy male volunteer. For spectra, the averaged values of the encoded 128 FIDs are used. Nonderivative \((m=0)\) and derivative \((m>0)\) magnitude spectra for the water-unsuppressed and water-suppressed zero-filled FID are on panels (a-d) and (e-h) in the left and right columns, respectively. The FFT spectra (a, e) with \(m=0\) are with no filtering. The optimized dFFT spectra (b-d; f-h) with the derivative orders \(1\le m\le 3\) are normalized and refer to the adaptive power-exponential filter, the APEF, for the damping parameter \(\alpha _1=3\). The spectral intensities on the ordinates are in arbitrary units (au). Resonance frequencies (chemical shifts) on the abscissae are in dimensionless units, parts per million (ppm). For details, see the text
Figure 3 is of the same configuration as Fig. 2. It also shows the spectra for the FIDs without and with water suppression on the left and right columns, respectively. At the common frequencies, the global features of the spectra for the APEF (Fig. 3) are quite similar to their counterparts for the APGF (Fig. 2). This occurs in spite of the difference between the damping parameters \(\alpha _2=5\) and \(\alpha _1=3\) from the APGF and APEF in Figs. 2 and 3, respectively. We recall that, even if \(\alpha _1\) and \(\alpha _2\) were the same, the smoothing parameters \(\lambda _1(m,T)\) in the APEF and \(\lambda _2(m,T)\) in the APGF would still be different, i.e. \(\lambda _2(m,T)=(1/T)\lambda _1(m,T)\) for \(\alpha _2=\alpha _1\).
As to the said similarities, the first derivative spectra with \(m=1\) (b) in Figs. 2 and 3 also shows the inverted peaks at 3.0-4.0 ppm for the same three resonances assigned to tCho, m-Ins and tCr. However, there are some differences between Figs. 2 and 3 for e.g. the third derivative spectra with \(m=3\) (d). These latter spectra are better resolved in Fig. 3 than in Fig. 2. In Fig. 3 too, the numbers on the ordinates differ even for the converged derivative spectra on panels (d) and (h) for the FIDs without and with water suppression, respectively. The parlance about this point goes in the same vein as with Fig. 2.
Focusing upon Fig. 3 alone, it appears that in some parts of the third derivative spectra with \(m=3\) (d, h), at e.g. 2.2-2.7 ppm and 3.30-4.25 ppm, the lineshapes are better resolved for the FIDs without than with water suppression. This is especially true for the m-Ins triplet (\(\sim \)4.07 ppm) and the NAA doublet (\(\sim \)4.17 ppm). Further, on panel (d), the tCr peak (\(\sim \)3.95 ppm) exhibits the shoulders on both side, as opposed to its broad structureless counterpart on panel (h). Moreover, the resonance assigned to glucose (Glc, \(\sim \)3.85 ppm) is seen on panel (d), but not on panel (h).
Within e.g. 2.2-2.7 ppm in Fig. 3, this favorable trend toward higher resolution for the FID without water suppression is initiated by the second derivative spectra with \(m=2\) (c). In this latter tight sub-band, unlike the situation in panel (g), some notches appear on panel (c) at the top of the peaks near 2.35 and 2.6 ppm for the Glx and NAA metabolites, respectively, marking the onset of separations of the overlapping resonances. This finding for \(\alpha _1=3\) in the APEF indicates that the procedure of water suppression in the course of the FID encodings is prone to introduce certain visible distortions in the spectra (e.g. the blur of the finer structures) even within the bands that are quite distant from the \(\mathrm{H_2O}\) chemical shift, 4.68 ppm.
In Fig. 4, we are back to the APGF in the optimized dFFT, which is for the water-unsuppressed FID alone. The derivative order is fixed at \(m=3\), whereas the Gaussian damping parameter \(\alpha _2\) is varied through some five values (1.75, 2.0, 2.25, 2.5, 5.0). Actually, the spectra in the optimized dFFT are plotted in the reverse orders (from 5.0 to 1.75) on panels (b-f), respectively. They are seen to undergo substantial alterations when \(\alpha _2\) is gradually reduced from 5.0 (b) to 1.75 (f). Herein, the spectrum on panel (b) for \(\alpha _2=5\) is sparse and populated by broad, rounded resonances first encountered in Fig. 2d. This intense smoothing is a direct consequence of the strong damping, \(\alpha _2=5\), which prevents the peak splitting.

Single-voxel in vivo proton MRS for white matter in the brain of a 25 year old healthy male volunteer. For spectra, the averaged value of the encoded 128 FIDs is used. Nonderivative \((m=0)\) and derivative \((m>0)\) magnitude spectra are for the water-unsuppressed zero-filled FID. The FFT spectrum (a) with \(m=0\) is with no filtering. The optimized dFFT (b-f) with the single derivative order \(m=3\) are normalized and refer to the adaptive power-Gaussian filter, the APGF, for five damping parameters \(\alpha _1=1.75, \,2.0, \,2.25, \, 2.5\) and 5.0. The spectral intensities on the ordinates are in arbitrary units (au). Resonance frequencies (chemical shifts) on the abscissae are in dimensionless units, parts per million (ppm). For details, see the text
However, by halving the value of the damping parameter \(\alpha _2\) to 2.5 (c), some refinements begin to develop through a partial unsmoothing in most of the spectrum, particularly at 2.3-2.9 ppm and 3.3-4.05 ppm. Importantly, another manifestation of the weaker damping (\(\alpha _2=2.5)\), with the ensuing diminished smoothing on panel (c), is the appearance of the additional five resonances. They are assigned to glucose (Glc, \(\sim \)3.85 ppm), m-Ins (\(\sim \)3.7 ppm), taurine (tau, \(\sim \)3.4 ppm), scyllo-inositol (s-Ins, \(\sim \)3.35 ppm) and aspartate (Asp, \(\sim \)2.8 ppm).
On the right panels, the damping parameter is sequentially and equidistantly reduced by a factor of 0.25 so as to have \(\alpha _2=2.25\) (d), \(\alpha _2=2.0\) (e) and \(\alpha _2=1.75\) (f). Peak delineations and the overall resolution is significantly improved on panel (d) relative to (c). This trend is pursued further on panels (e) and (f). They both propel two additional peak assigned to glutathione (GSH, \(\sim \)2.9 ppm) and nitrogen acetyl aspartyl glutamic acid (NAAG, \(\sim \)2.1 ppm). On panel (e), the NAAG peak begins its slight separation, which eventually becomes more pronounced on panel (f). Moreover, the latter panel is supplemented by an extra resonance, assigned to gamma amino butyric acid (GABA, \(\sim \)1.9 ppm).

Single-voxel in vivo proton MRS for white matter in the brain of a 25 year old healthy male volunteer. For spectra, the averaged value of the encoded 128 FIDs is used. Nonderivative \((m=0)\) and derivative \((m>0)\) magnitude spectra are for the water-suppressed zero-filled FID. The FFT spectra with \(m=0\) in the quasi-absorptive (a) and magnitude (e) modes are with no filtering. The real part (a) of the complex FFT spectrum is for the complex zero-order phase-corrected FID, which is multiplied by \(\textrm{e}^{i\varphi _0}\), where \(\varphi _0=80^\circ \) (or 1.3963 rad). The optimized dFFT spectra (b-d; f-h) in the magnitude mode alone with the derivative orders \(1\le m\le 3\) refer to the adaptive power-exponential filter, the APEF, for two damping parameters \(\alpha _1=1.5\) and 3.0. The spectral intensities on the ordinates are in arbitrary units (au). Resonance frequencies (chemical shifts) on the abscissae are in dimensionless units, parts per million (ppm). For details, see the text
It is then seen that the usage of a fixed derivative order (\(m=3\)) in conjunction with the varying Gaussian damping parameter \(\alpha _2\) proves helpful. It offers an opportunity to monitor the pattern of the gradual, systematic and simultaneous improvement of resolution and SNR by the optimized dFFT for the FIDs encoded without water suppression. Such an outcome gains in its importance especially when juxtaposed to the FFT (a), which provides no meaningful information. The optimized dFFT makes a notable progress by fine-tuning the smoothing parameter \(\alpha _2\). This results in an almost doubling of the number of the identified physical resonances K when decreasing \(\alpha _2\) by a factor of 2.857 during the passage from \(\alpha _2=5\) (b), yielding \(K=11\), to \(\alpha _2=1.75\) (f), giving \(K=20\).
Monitoring the relative stabilization of the derivative spectra from the optimized dFFT should likewise be instructive for the water-suppressed FIDs. This is illustrated Fig. 5, which differs from the preceding figure in several aspects. It uses the APEF (exponential damping) instead of the APGF (Gaussian damping). It runs through the derivative orders from one to three as opposed to having a single fixed values of m. It employs two damping parameters. In Fig. 5, the selected exponential damping parameters are \(\alpha _1=3\) (b-d) and \(\alpha _1=1.5\) (f-h).
The FFT spectra on panels (a) and (e) are in the quasi-absorptive and magnitude modes, respectively. On panel (a), the real part of the complex FFT spectrum is for the FIDs multiplied by \(\textrm{e}^{i\varphi _0}\), where \(\varphi _0=80^\circ \) (1.3963 rad). Such a phasing of the FID reduces the baseline which, in turn, allows a more identifiable tCr peak on panel (a) near 3.95 ppm than on panel (e). However, the spectrum on panel (a) is not positive-definite everywhere within 1.75-4.05 ppm. Therein, some of the inverted peaks have their negative intensities, as visible near 2.8 and 2.48 ppm. It then appears that the quasi-absorptive spectrum (a) is not of much help in practice. This finding echoes the already discussed unfavorable status of the magnitude envelope (e).
On the other hand, the derivative spectra from the optimized dFFT (b-d, f-h) in Fig. 5 possess a richer informational content than that from the FFT (a, e). This holds true for both spectral sets (b-d) and (f-h) with \(\alpha _1=3\) and \(\alpha _1=1.5\), respectively. With the forced stronger damping of the encoded FID by using \(\alpha _1=3\), the three spectra (\(1\le m\le 3\)) have their relatively stable lineshapes (b-d). Herein, some improvements can be observed by going from \(m=1\) (b) to \(m=3\) (d), especially with the dip between the tCr (\(\sim \)3.0 ppm) and tCho (\(\sim \)3.2 ppm) peaks as well as with the delineation of the Glx peak (\(\sim \)2.15ppm). Otherwise, for \(m=1\) (b), \(m=2\) (c) and \(m=3\) (d), the number of the identified metabolites stays put at \(K=12\) for \(\alpha _1=3\).
However, for the twice weaker smoothing (\(\alpha _1=1.5\)), there are more pronounced changes in the derivative profiles when passing from \(m=1\) (f), through \(m=2\) (g) to \(m=3\) (h). This is reflected in more splittings of the overlapped peaks, as seen in \(K=15\) (f) compared to \(K=19\) (g, h). For \(m=1\) (f), the tCr peak (\(\sim \)3.95 ppm) is not deconvolved into Cr and PCr. Moreover, the Glc peak (\(\sim \)3.85 ppm) is unidentifiable for \(m=1\) (f). Further, only the slight shoulders appear for \(m=1\) (f) at the expected peak locations assignable to the NAAG (\(\sim \)2.1 ppm) and GABA (\(\sim \)1.9 ppm) metabolites.
All these four deficiencies for \(m=1\) (f) are rectified either in part or with a fuller extent for \(m=2\) (g) and \(m=3\) (h), respectively. For example, the most pronounced improvements for \(m=3\) (h) are for the NAAG (\(\sim \)2.1 ppm) and GABA (\(\sim \)1.9 ppm) peaks. These two resonances are remarkably well separated from the dominant NAA peak (\(\sim \)2.0 ppm) all the way down to the chemical shift axis. Thus, in the optimized dFFT, compared to the left column (b-d) for \(\alpha _1=3\), the right column (f-h) for \(\alpha _1=1.5\) is manifestly superior regarding both resolution and SNR.
As emphasized, Figs. 4 and 5 are for the water-unsuppressed and water-suppressed FIDs, respectively. The main finding from the panel (h) in Figs. 4 and 5 is that the relatively stabilized derivative spectra for \(m=3\) are of comparably high-resolution and low noise. This is achieved with two different filters, the APGF (Fig. 4) and APEF (Fig. 5) using the smallest among several considered values of the damping parameters, i.e. \(\alpha _2=1.75\) (Gaussian attenuation) and \(\alpha _1=1.5\) (exponential attenuation).
It is gratifying that the derivative spectra from the optimized dFFT using the FIDs encoded by in vivo proton MRS with and without water suppression are of a similar excellent quality. This should be of particular importance in practice because of the possibility to significantly reduce the patient examination time which, in turn, could make MRS more cost-effective in the clinic.
In practice, e.g. the GE MRS protocol allows encodings of a collection of water-unsuppressed FIDs to have the reference data. Subsequently, after water suppression (by e.g. inversion recovery or selective excitation), the water-suppressed FIDs are acquired. Obviously, it would be hugely advantageous (for the patient as well as for in vivo MRS as a whole) if the water-unsuppressed FIDs could provide spectra of clinical reliability comparable to those from water-suppressed FIDs. As shown here, such a goal is untenable with the FFT, but it is achievable with the optimized dFFT.
4 Conclusions
The usual unattenuated derivative fast Fourier transform (dFFT) completely breaks down for measured time signals. These include free induction decay (FID) data encoded by in vitro or in vivo magnetic resonance spectroscopy (MRS). Such time signals are invariably dominated by noise at their tails, i.e. toward the end of the total acquisition time. The failure of the unattenuated dFFT in this case is attributed to multiplication of the given encoded FID by a time power function (a monomial) stemming from the frequency derivative operators applied to the unattenuated fast Fourier transform (FFT). This eliminates the possibility for splitting the overlapping spectral resonances and, moreover, it worsens the signal-to-noise ratio (SNR).
A solution to this problem is provided by an adaptive optimization of the unattenuated dFFT. This is accomplished by means of the attenuated exponential or Gaussian filters. Each of these is completely specified by a single damping parameter derived in the simple analytical forms. These universally applicable optimal filters are simultaneously tailored to the derivative order, frequency resolution and SNR of the FID. The damping parameter is not a constant, but rather it is updated by being appropriately scaled with the derivative order. The virtues of the ensuing optimized dFFT for the phase-insensitive magnitude-mode spectra are:
-
automated application,
-
user-independence,
-
fast computation by the Cooley-Tukey algorithm,
-
no modeling of the FIDs nor of the spectra,
-
symmetrized lineshapes,
-
narrowed widths of metabolite peaks (the smallest full widths at half maximae),
-
simultaneously enhanced resolution as well as SNR,
-
absence of lineshape distortions for increasing derivative orders,
-
suppression of sidelobes, and
-
background reduction.
The listed beneficial features are of relevance to the field of signal processing, in general, and to medical diagnostics by MRS, in particular, all marred by the common ‘identification problem’. This problem refers to the accurate and reliable determination of the individual components hidden (folded, masked) within the overlapped spectral bands. By reliance upon shape estimation alone, the optimized dFFT can decompose the given FFT envelope (total shape spectrum) into its true non-overlapping partial lineshapes of resonances assignable to individual constituents of the investigated specimen and amenable to straightforward quantification by e.g. integration.
It is the adaptive power-exponential filter (APEF) or the adaptive power-Gaussian filter (APGF) in the magnitude spectra from the optimized dFFT that tightens the peak base, narrows the peak width, increases the peak height, enhances resolution and suppresses noise, background and the peak sidelobes. As opposed to the magnitude lineshapes, in the ‘absorption’ mode spectra, the derivative peak sidelobe intensities increase with the augmented derivative order and can be misinterpreted as physical resonances. The tightened peak base translates into the lineshape localization to a very narrow range around the genuine resonance frequency. In particular, for the water resonance, this amounts to a veritable water suppression. The net result from this novel water suppression device based on the optimized dFFT is the emergence of the other peaks assignable to metabolites of diagnostic relevance. Formerly, these latter peaks were hidden in the macromolecular background superimposed on the long and elevated water tails.
The presently reported spectra from the optimized dFFT are for short FIDs (0.5 KB), encoded at a low-field clinical scanner (1.5T) using a long echo time (272 ms) with and without water suppression by proton MRS from white matter in the brain of a 25 year old healthy male volunteer. For both extremely different FIDs, this processor reconstructs spectra of similarly high resolution and low noise. This shows that proton MRS at 1.5T clinical scanners can provide fully respectable FIDs and it is only the matter of employing an adequate estimator, such as the optimized dFFT, for appropriate signal processing. Encoding the water-unsuppressed FIDs alone can considerably shorten the patient examination time and contribute to significantly improved cost-effectiveness of MRS as a necessary requirement for becoming a standard everyday diagnostic modality in the clinic.
Data availability
Data from this work can be made available to other researchers in this field upon request to the Authors.
Notes
Note that the same capital letter, but in the Roman and Italic fonts, refers to different quantities: T for the unit of tesla and T for the total acquisition time.
In Ref. [68], there were two misprints that are corrected here as: (i) \(\textrm{e}^{-3}\approx 0.4598\) should read \(\textrm{e}^{-3}\approx 0.0498\) and (ii) \(t^m\) amplies noise at larger t should read \(\longrightarrow \) \(t^m\) amplifies noise at larger t. The reconstructions from Ref. [68] were not influenced by the misprint (i) since the optimized dFFT employed the correct approximate value 0.0498 for \(\textrm{e}^{-3}\).
References
I.I. Rabi, J.R. Zacharias, S. Millman, P. Kusch, A new method of measuring nuclear magnetic moment. Phys. Rev. 53, 318 (1938)
E.M. Purcell, H.C. Torrey, R.V. Pound, Resonance absorption by nuclear moments in a solid. Phys. Rev. 69, 37–38 (1946)
F. Bloch, W.W. Hansen, M. Packard, Nuclear induction. Phys. Rev. 69, 127 (1946)
K. Siegbahn, G. Lindström, Magnetic moments of deuterium-2, lithium-7 and fluorine-19. Nature 163, 211 (1949)
W.D. Knight, Nuclear magnetic resonance shift in metals. Phys. Rev. 76, 1259–1260 (1949)
W.G. Proctor, F.C. Yu, The dependence of a nuclear magnetic resonance frequency upon chemical compound. Phys. Rev. 77, 717 (1950)
H.C. Gutowsky, R.E. McClure, Magnetic shielding of the proton resonance in \({\rm H_2, H_2O}\) and mineral oil. Phys. Rev. 81, 276–277 (1951)
E.L. Hahn, Spin echoes. Phys. Rev. 80, 580–594 (1950)
E.R. Danielsen, B. Ross, Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases (Marcel Dekker, New York, 1999)
L.A. Brandão, R.C. Dominges, MR Spectroscopy of the Brain (Lippincott Williams & Wilkins, Philadelphia, 2004)
Dž. Belkić, K. Belkić, eds., Magnetic Resonance Imaging and Spectroscopy, Vol. 3, In: Comprehensive Biomedical Physics (10 Volumes) (Elsevier, Amsterdam, 2014)
J.R. Griffiths, P.A. Bottomley, Handbook of Magnetic Resonance Spectroscopy In Vivo: MRS Theory, Practice and Application (John Wiley & Sons, 2016)
R.A. de Graaf, In Vivo NMR Spectroscopy: Principles and Techniques, 3rd edn. (John Wiley & Sons, Chilchester, 2018)
G. Öz, J.R. Alger, P.B. Barker, R. Bartha, A. Bizzi, C. Boesch, P.J. Bolan, K.M. Brindle, C. Cudalbu, A. Dinçer, U. Dydak, U.E. Emir, J. Frahm, R.G. González, S. Gruber, R. Gruetter, R.K. Gupta, A. Heerschap, A. Henning, H.P. Hetherington, F.A. Howe, P.S. Hüppi, R.E. Hurd, K. Kantarci, D.W.J. Klomp, R. Kreis, M.J. Kruiskamp, M.O. Leach, A.P. Lin, P.R. Luijten, M. Marjańska, A.A. Maudsley, D.J. Meyerhoff, C.E. Mountford, S.J. Nelson, M.N. Pamir, J.W. Pan, A.C. Peet, H. Poptani, S. Posse, P.J.W. Pouwels, E.-M. Ratai, B.D. Ross, T.W.J. Scheenen, C. Schuster, I.C.P. Smith, B.J. Soher, I. Tkáč, D.B. Vigneron, R.A. Kauppinen, Clinical proton MR spectroscopy in central nervous system disorders. Radiology 270, 658–679 (2014)
M. Wilson, O. Andronesi, P.B. Barker, R. Bartha, A. Bizzi, P.J. Bolan, K.M. Brindle, I.-Y. Choi, C. Cudalbu, U. Dydak, U.E. Emir, R.G. González, R. Gruber, R. Gruetter, R.K. Gupta, A. Heerschap, A. Henning, H.P. Hetherington, P.S. Huppi, R.E. Hurd, K. Kantarci, R.A. Kauppinen, D.W.J. Klomp, R. Kreis, M.J. Kruiskamp, M.O. Leach, A.P. Lin, P.R. Luijten, M. Marjańska, A.A. Maudsley, D.J. Meyerhoff, C.E. Mountford, P.G. Mullins, J.B. Murdoch, S.J. Nelson, R. Noeske, G. Öz, J.W. Pan, A.C. Peet, H. Poptani, S. Posse, E.-M. Ratai, N. Salibi, T.W.J. Scheenen, I.C.P. Smith, B.J. Soher, I. Tkáč, D.B. Vigneron, F.A. Howe, Methodological consensus on clinical proton MRS of the brain: Review and recommendations. Magn. Reson. Med. 82, 527–550 (2019)
C. Cudalbu, K.L. Behar, P.K. Bhattacharyya, W. Bogner, T. Borbath, R.A. de Graaf, R. Gruetter, A. Henning, C. Juchem, R. Kreis, Contribution of macromolecules to brain \({}^1{\rm H}\) MR spectra: Experts’ consensus and recommendations. NMR Biomed. 34, e4393 (2021)
I. Tkáč, D. Deelchand, W. Dreher, H. Hetherington, R. Kreis, C. Kumaragamage, M. Považan, D.M. Spielman, B. Strasser, R.A. de Graaf, Water and lipid suppression techniques for advanced \({}^1{\rm H}\) MRS and MRSI of the human brain: Experts’ consensus recommendations. NMR Biomed. 34, e4459 (2021)
E. Odeblad, G. Lindström, Some preliminary observations on the proton magnetic resonances in biological samples. Acta Radiol. 43, 469–476 (1955)
P.T. Beall, A historical perspective on biomedical NMR. Magn. Reson. Imag. 1, 189-190 (1982) Editorial Comment on Ref. [18]
R.E. Ernst, W.A. Anderson, Application of Fourier transform spectroscopy to magnetic resonance. Rev. Sci. Instrum. 37, 93–102 (1966)
J. Frahm, K.D. Merboldt, W. Hänicke, Localized proton spectroscopy using stimulated echoes. J. Magn. Reson. 72, 502–508 (1987)
P.A. Bottomley, Spatial localization in NMR spectroscopy in vivo. Ann. N.Y. Acad. Sci. 508, 333–348 (1987)
J. Hennig, H. Pfister, T. Ernst, O. Ott, Direct absolute quantification of metabolites in the human brain with in vivo localized proton spectroscopy. NMR Biomed. 5, 193–199 (1992)
J.R. Alger, S.C. Symko, A. Bizzi, S. Posse, D.J. DesPres, M.R. Armstrong, Absolute quantification of short TE brain \({}^1{\rm H}-\)MR spectra and spectroscopic imaging data. J. Comput. Assist. Tomogr. 17, 191–199 (1993)
B. Ross, T. Michaelis, Clinical applications of magnetic resonance spectroscopy. Magn. Reson. Quarterly 10, 191–247 (1994)
T. Schirmer, D.P. Auer, On the reliability of quantitative clinical magnetic resonance spectroscopy of the human brain. NMR Biomed. 13, 28–36 (2000)
I. Tkáč, P. Andersen, G. Adriany, H. Merkle, K. Uǧurbil, R. Gruetter, In vivo \({}^1{\rm H}\) NMR spectroscopy of the human brain at 7T. Magn. Reson. Med. 46, 451–456 (2001)
D.J. Drost, W.R. Riddle, G.D. Clarke, Proton magnetic resonance spectroscopy in the brain: Report of AAPM MR Task Group # 9. Med. Phys. 29, 2177–2197 (2002)
F.A. Howe, S.J. Barton, S.A. Cudlip, M. Stubbs, D.E. Saunders, M. Murphy, P. Wilkins, K.S. Opstad, V.L. Doyle, M.A. McLean, B.A. Bell, J.R. Griffiths, Metabolic profiles of human brain tumors using quantitative in vivo \({}^1{\rm H}\) magnetic resonance spectroscopy. Magn. Reson. Med. 49, 223–232 (2003)
J.J. Geurts, F. Barkhof, J.A. Castelijns, B.M. Uitdehaag, C.H. Polman, P.J. Pouwels, Quantitative \({}^1{\rm H}\)-MRS of healthy human cortex, hippocampus, and thalamus: Metabolite concentrations, quantification precision, and reproducibility. J. Magn. Reson. Imag. 20, 366–371 (2004)
S. Ramadan, M.A. Thomas, C.E. Mountford, Phase-rotated MR spectroscopy using dual-PRESS: Theory and applications in human brain. AIP Conf. Proc. 953, 277–291 (2007)
S. Gruber, K. Pinker, F. Riederer, M. Chmelik, A. Stadlbauer, M. Bittsanský, V. Mlynárik, R. Frey, W. Serles, O. Bodamer, E. Moser, Metabolic changes in the normal aging brain: Consistent findings from short and long echo time proton spectroscopy. Eur. J. Radiol. 68, 320–327 (2008)
A. Horská, P.B. Barker, Imaging of brain tumors: MR spectroscopy and metabolic imaging. Neuroimag. Clin. N. Am. 20, 293–310 (2010)
J.R. Alger, Quantitative proton magnetic resonance spectroscopy and spectroscopic imaging of the brain: A didactic review. Top. Magn. Reson. Imag. 21, 115–128 (2010)
I. Ronen, E. Ercan, A. Webb, Rapid multi-echo measurement of brain metabolite \(T_2\) values at 7T using single-shot spectroscopic Carr-Purcell-Meiboom-Gill sequence and prior information. NMR Biomed. 26, 1292–1298 (2013)
M.H. Buonocore, R.J. Maddock, Magnetic resonance spectroscopy of the brain: A review of physical principles and technical methods. Rev. Neurosci. 26, 609–632 (2015)
M.C. Munõz-Hernández, M.L. García-Martín, In vivo \({}^1{\rm H}\) magnetic resonance spectroscopy, Vol. 1718, pp. 151-167, In: M. García-Martín, P. López-Larrubia, Preclinical MRI: Methods in Molecular Biology (Humana Press, New York, 2018)
W. Dreher, D. Leibfritz, New method for the simultaneous detection of metabolites and water in localized in vivo 1H nuclear magnetic resonance spectroscopy. Magn. Reson. Med. 54, 190–195 (2005)
Z. Dong, Proton MRS and MRSI of the brain without water suppression. Prog. Nucl. Magn. Reson. Spectrosc. 86/87, 65–79 (2015)
U.E. Emir, B. Burns, M. Chiew, P. Jezzard, M.A. Thomas, Non-water-suppressed short-echo-time magnetic resonance spectroscopic imaging using a concentric ring \(k-\)space trajectory. NMR Biomed. 30, e3714 (2017)
J. Dorst, T. Borbath, K. Landheer, N. Avdievich, A. Henning, Simultaneous detection of metabolite concentration changes, water BOLD signal and pH changes during visual stimulation in the human brain at 9.4T. J. Cereb. Blood Flow Metab. 46, 1104–1119 (2022)
V. Govindaraju, K. Young, A.A. Maudsley, Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed 13, 129–153 (2000)
Dž. Belkić, Fast Padé Transform (FPT) for magnetic resonance imaging and computerized tomography. Nucl. Instrum. Meth. Phys. Res. A 471, 165–169 (2001)
Dž. Belkić, K. Belkić, Fast Padé transform for optimal quantification of time signals from magnetic resonance spectroscopy. Int. J. Quantum Chem 105, 493–510 (2005)
Dž. Belkić, Quantum-Mechanical Signal Processing and Spectral Analysis (Taylor & Francis, London, 2005)
Dž. Belkić, K. Belkić, Signal Processing in Magnetic Resonance Spectroscopy with Biomedical Applications (Taylor & Francis, London, 2010)
Dž. Belkić, K. Belkić, Quantification by the fast Padé transform of magnetic resonance spectroscopic data encoded at 1.5T: Implications for brain tumor diagnostics. J. Math. Chem. 54, 602–655 (2016)
M. Gajdošík, K. Landheer, K.M. Swanberg, C. Juchem, INSPECTOR: free software for magnetic resonance spectroscopy data inspection, processing, simulation and analysis. Sci. Rep. 11, 2094 (2021) [The open-source freely downloadable complete package (since 2017) at: http://innovation.columbia.edu/technologies/cu17130l_inspector]
W.T. Clarke, C.J. Stagg, S. Jbabdi, FSL-MRS: An end-to-end- spectroscopy analysis package. Magn. Reson. Med. 85, 2950–2964 (2021) [The open-source freely downloadable complete package at: http://www.git.fmrib.ox.ac.uk/fsl/fsl_mrs]
A. Schrantee, C. Najac, C. Jungerius, W. van der Zwaag, S. Jbabdi, W.T. Clarke, I. Ronen, A 7T interleaved fMRS and fMRI study on visual contrast dependency in the human brain. Imag. Neurosci. 1 (2023) [https://doi.org/10.1162/imag_e_00007]
J.C. Lindon, A.G. Ferrig, Digitisation and data processing in Fourier transform NMR. Progr. NMR Spectr. 14, 27–66 (1980)
R.N. Bracewell, The Fourier Transform and Its Applications, 3rd edn. (McGraw-Hill, New York, 2000)
Dž. Belkić, K. Belkić, Exact quantification by the nonparametric fast Padé transform using only shape estimation of high-order derivatives of envelopes. J. Math. Chem 56, 268–314 (2018)
Dž. Belkić, K. Belkić, Explicit extraction of absorption peak positions, widths and heights using higher order derivatives of total shape spectra by nonparametric processing of time signals as complex damped multi-exponentials. J. Math. Chem. 56, 932–977 (2018)
Dž. Belkić, K. Belkić, Validation of reconstructed component spectra from non-parametric derivative envelopes: Comparison with component lineshapes from parametric derivative estimations with the solved quantification problem. J. Math. Chem. 56, 2537–2578 (2018)
Dž. Belkić, K. Belkić, Review of recent applications of the conventional and derivative fast Padé transform for magnetic resonance spectroscopy. J. Math. Chem. 57, 385–464 (2019)
Dž. Belkić, K. Belkić, Feasibility study for applying the lower-order derivative fast Padé transform to measured time signals. J. Math. Chem. 58, 146–177 (2020)
Dž. Belkić, K. Belkić, Derivative NMR spectroscopy for J-coupled multiplet resonances with short time signals (0.5KB) encoded at low magnetic field strengths (1.5T): Part I, Water Suppressed. J. Math. Chem. 59, 364–404 (2021)
Dž. Belkić, K. Belkić, Derivative NMR spectroscopy for J-coupled multiplet resonances with short time signals (0.5KB) encoded at low magnetic field strengths (1.5T): Part II, Water Unsuppressed. J. Math. Chem. 59, 405–443 (2021)
Dž. Belkić, K. Belkić, In vivo derivative NMR spectroscopy for simultaneous improvements of resolution and signal-to-noise-ratio: Case study. Glioma. J. Math. Chem 59, 2133–2178 (2021)
Dž. Belkić, K. Belkić, High-resolution at 3T for in vivo derivative NMR spectroscopy in medical diagnostics of ovarian tumor: Exact quantification by shape estimations. J. Math. Chem. 59, 2218–2260 (2021)
Dž. Belkić, K. Belkić, Derivative NMR spectroscopy for J-coupled resonances in analytical chemistry and medical diagnostics. Adv. Quantum Chem. 84, 95–265 (2021)
Dž. Belkić, K. Belkić, In vitro proton magnetic resonance spectroscopy at 14T for benign and malignant ovary: Part I, Signal processing by the nonparametric fast Padé transform. J. Math. Chem. 60, 373–416 (2022)
Dž. Belkić, K. Belkić, In vitro proton magnetic resonance spectroscopy at 14T for benign and malignant ovary: Part II, Signal processing by the parametric fast Padé transform. J. Math. Chem. 60, 1200–1271 (2022)
Dž. Belkić, K. Belkić, Magnetic resonance spectroscopy at high magnetic fields: Derivative reconstructions of components from envelopes using encoded time signals. Adv. Quantum Chem. 86, 151–221 (2022)
Dž. Belkić, K. Belkić, Inverse problem for reconstruction of components from derivative envelope in ovarian MRS: Citrate quartet as a cancer biomarker with considerably decreased levels in malignant vs benign samples. J. Math. Chem. 61, 569–599 (2023)
Dž. Belkić, K. Belkić, Derivative shape estimations with resolved overlapped peaks and reduced noise for time signals encoded by NMR spectroscopy with and without water suppression. J. Math. Chem. 61, 1936–1966 (2023)
Dž. Belkić, K. Belkić, Optimized derivative fast Fourier transform with high resolution and low noise from encoded time signals: Ovarian NMR spectroscopy. J. Math. Chem. (2023), 62, 535–554 (2024)
J.W. Cooley, J.W. Tukey, An algorithm for machine calculation of complex Fourier series. Math. Comp. 19, 297–301 (1965)
Manual, Spectroscopy Application Guide Gyroscan ACS-NT (Philips Medical System Nederland BV, 1989)
E.A. Boss, S.H. Moolenaar, L.F. Massuger, H. Boonstra, U.F. Engelke, J.G. de Jong, R.A. Wevers, High-resolution proton nuclear magnetic resonance spectroscopy of ovarian cyst fluid. NMR Biomed. 13, 297–305 (2000)
Acknowledgements
The authors thank the ‘Radiumhemmet Research Fund’ at the Karolinska University Hospital, the ‘Fund for Research and Development & Education (FoUU) of the Stockholm County Council’. Open Access has been provided by the Karolinska Institute, Stockholm, Sweden.
Funding
Open access funding provided by Karolinska Institute. Open access funding provided by the Karolinska Institute. This work is funded by the Radiumhemmet Research Funds, King Gustaf the Fifth Jubilee Fund at the Karolinska University Hospital, Fund for Research, Development and Education (FoUU) of the Stockholm County Council.
Author information
Authors and Affiliations
Contributions
Signal processing and the art work have been carried out by DžB. Analysis of the obtained spectra and contributions to writing with checking the texts have been performed by DžB and KB. Both authors cooperatively designed this study and critically read as well as approved the final version of the manuscript submitted for publication.
Corresponding author
Ethics declarations
Conflict of interest
The Authors have no competing interests.
Ethical approval
The Regional Ethics Committee, Karolinska Institute, Stockholm (DNR # 708-31/1, Protocol 20-06-2007) found no ethical issues that would preclude carrying out this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belkić, D., Belkić, K. In vivo brain MRS at a 1.5T clinical scanner: Optimized derivative fast Fourier transform for high-resolution spectra from time signals encoded with and without water suppression. J Math Chem 62, 1251–1286 (2024). https://doi.org/10.1007/s10910-024-01597-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10910-024-01597-6