
Abstract
Pyrazoline and pyrazole compounds are important building blocks in the development of photofunctional compounds. Two new solution-processable platinum-containing polyynes functionalized with these chromophores in the main chain (P1 and P2) were synthesized and characterized by spectroscopic, thermal and optical methods. By changing the spacer from 2-pyrazoline to the electron-rich pyrazole in the main chain, the photophysical properties including energy levels, absorption wavelength and bandgap of the polymers were finely tuned. The photovoltaic properties of P1 with better absorption features were also studied.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The development of transition metal polyynes of the form trans-[−M(L)2-C≡C-Ar-C≡C−]n (L = auxiliary ligands, Ar = spacer unit) has shown much progress in the past few decades [1,2,3,4,5]. These main-chain organometallic polymers are useful functional materials for luminescence, photovoltaics, biology, nonlinear optics (NLO) and chemosensing [1,2,3,4,5,6]. To date, a large family of soluble platinum(II) polyynes incorporating various conjugated carbocyclic and heteroaromatic ring systems is known [7,8,9,10,11,12].
2-Pyrazoline derivatives are good candidates for constructing versatile optical materials [13,14,15,16]. These compounds show fluorescence and electrooptical properties and by means of easy synthetic procedures, allow the introduction of appropriate substituents for tailoring the second-order NLO properties. In particular, 1,3,5-triaryl-2-pyrazoline derivatives have been studied due to their high photoluminescence and good electrochemical activity and are widely applied as fluorescent probes, NLO materials and OLED materials [17]. Pyrazoline has an interesting ring containing both an electron-accepting imine –C = N– group and an electron-donating –N(Ar)– group. The N atom of pyrazoline attains conjugation by electron donation which leads to higher hole-transport efficiency and some photoelectron characteristics [13]. Therefore, the pyrazoline derivatives are expected to be used as a new type of optoelectronic materials in a broader field of applications. On the other hand, the electron-rich pyrazoles are an important class of nitrogen-containing five-membered heterocycles and show a range of applications in medicine, dye-sensitized solar cells and second-order NLO [18,19,20,21,22,23,24,25]. To our knowledge, there are no reports on exploiting pyrazoline and its pyrazole derivatives in the preparation of organometallic polyynes. In this work, two new polyplatinayne polymers (P1 and P2) were designed and synthesized, which contain structurally similar pyrazoline and pyrazole rings in the polymer main chain.
2 Experimental Section
2.1 Materials and Instruments
All the chemicals were purchased from Acros or Aldrich and used as received unless otherwise specified. trans-[Pt(PBu3)2Cl2] [26] was prepared using the methods reported in the literature. THF was dried by distillation from sodium with benzophenone as an indicator under a nitrogen atmosphere. Separation and purification of products were achieved by column chromatography on silica gel. TLC was carried out in air using laboratory grade solvents as eluents.
The positive-ion fast atom bombardment (FAB) mass spectra were recorded in m-nitrobenzyl alcohol matrices on a Finngin-MAT SSQ710 mass spectrometer. Infrared spectra were recorded on the Nicolet Magna 550 Series II FTIR spectrometer using KBr pellets for solid state spectroscopy. NMR spectra were measured in deuterated solvents as the lock and reference on Varian INOVA 400 instrument or Bruker AV 400 MHz FT-NMR spectrometer, with 1H and 13C NMR chemical shifts quoted relative to Me4Si standard and 31P chemical shifts relative to an 85% H3PO4 external reference. Electronic absorption spectra were obtained with a Hewlett Packard 8453 spectrometer. Solution state photoluminescence measurements were obtained by the LS50B fluorescent spectrometer. For lifetime measurements, the third harmonics, 355 nm line of a Q-switched Nd: YAG laser was used as the excitation light source. The emission was recorded by using a PMT and an HP54522A 500 MHz oscilloscope. The PL spectra were measured in CH2Cl2 with a PTI Fluorescence Master Series QM1 spectrophotometer. The molecular weights of the polymers were determined by gel permeation chromatography (GPC) (HP 1050 series HPLC with visible wavelength and fluorescent detectors) using polystyrene standards. Thermal analysis was performed with a Perkin-Elmer TGA6 thermal analyzer.
The cyclic voltammograms were acquired with a CHI model 600D electrochemical station in deoxygenated acetonitrile containing 0.1 M [Bu4N]PF6 as the supporting electrolyte. A conventional three-electrode configuration consisting of a platinum working electrode, a Pt-wire counter electrode and an Ag/AgCl reference electrode was used. The polymer films were cast on the ITO covered glass. All potentials reported were quoted with reference to the ferrocene-ferrocenium (Fc/Fc+) couple at a scan rate of 100 mV s− 1.
2.2 Fabrication and Characterization of Polymer Solar Cells
PCBM was purchased from American Dyes while PEDOT/PSS (Baytron P VPAI 4083) was purchased from H. C. Starck. Device configuration of ITO/poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS)/polymer:PCBM/Al was applied in this study. Indium tin oxide (ITO) coated glass substrates (10 Ω per square) were cleaned by sonication in toluene, acetone, ethanol, and deionized water, dried in an oven, and then cleaned with UV ozone for 300 s. As-received PEDOT: PSS solution was passed through the 0.45 µm filter and spin-coated on patterned ITO substrates at 5000 rpm for 3 min, followed by baking in N2 at 150 °C for 15 min. P1:PCBM (1:4 by weight) active layer was prepared by spin-coating the toluene solution (4 mg mL–1 of P1 and 16 mg mL–1 of PCBM) at 800 rpm for 2 min. The substrates were dried at room temperature under vacuum for 1 h and then stored under high vacuum (10–5 to 10–6 Torr) overnight. An Al electrode (100 nm) was evaporated through a shadow mask to define the active area of the devices (2 mm2 circle). All the fabrication procedures (except drying, PEDOT:PSS annealing and Al deposition) and cell characterization were performed in air. Power conversion efficiency (PCE) was determined from J–V curve measurement (using a Keithley 2400 sourcemeter) under white light illumination (at 100 mW cm–1). For white light efficiency measurements, an Oriel 66,002 solar light simulator with an AM1.5 filter was used. The light intensity was measured by a Molectron Power Max 500D laser power meter. For the measurement of the external quantum efficiency (EQE), different wavelengths were selected with an Oriel Cornerstone 74,000 monochromator, while the photocurrent was measured with a Keithley 2400 sourcemeter. The light intensity was measured with a Newport 1830-C optical power meter equipped with an 818-UV detector probe.
2.3 General Procedures for the Synthesis of Polyplatinayne Polymers P1 and P2
2.3.1 Synthesis of 1-(4-bromophenyl)-3-phenylprop-2-en-1-one
In 100 mL round-bottom flask, benzaldehyde (1.2 mL, 11.80 mol) was dissolved in absolute alcohol (32 mL) at room temperature. After 2.5 M NaOH (32 mL) was added, 1-(4-bromophenyl)ethanone (2.40 g, 12.06 mmol) was added and the resulting mixture was stirred for 3 h at room temperature. The precipitate was collected, washed with ice-cooled water and recrystallized from ethanol and dried to obtain 1-(4-bromophenyl)-3-phenylprop-2-en-1-one (3.40 g, 11.84 mmol, 88%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.91‒7.89 (m, 2 H, Ar), 7.83 (m, 1H, Ar), 7.67‒7.64 (m, 4 H, Ar), 7.51‒7.46 (m, 1H, CH = CH), 7.44 (m, 3 H, Ar + CH = CH) ppm. 13C NMR (100 MHz, CDCl3): δ = 189.43 (C = O), 145.47, 136.89, 134.65, 131.96, 130.81, 129.04, 128.55, 127.94, 121.42 (Ar + C = C) ppm. FAB-MS: m/z = 289.1 [M]+.
2.3.2 Synthesis of 1,3-bis(4-bromophenyl)-5-phenyl-4,5-dihydro-1H-pyrazole L1-Br
A mixture of 1-(4-bromophenyl)-3-phenylprop-2-en-1-one (1.63 g, 5.66 mmol) and 1-bromophenylhydrazine hydrochloride (1.27 g, 5.66 mmol) in 20 mL ethanol was heated to reflux for 24 h under a nitrogen atmosphere. After the mixture was cooled down to room temperature, the precipitate was collected and washed with ethanol and dried to obtain L1-Br (2.30 g, 5.05 mmol, 89%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.58‒7.56 (m, 2 H, Ar), 7.52‒7.49 (m, 2 H, Ar), 7.34‒7.32 (m, 2 H, Ar), 7.29‒7.27 (m, 2 H, Ar), 7.26‒7.24 (m, shielded by CHCl3 proton, 2 H, Ar), 6.93‒6.90 (m, 2 H, Ar), 5.28‒5.24 (m, 1H, CH), 3.86‒3.78 (m, 1H, CH2), 3.15‒3.09 (m, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ = 146.25, 143.37. 141.68, 131.75, 131.71, 131.38, 129.31, 127.89, 127.20, 125.75, 122.82, 114.95, 111.40 (Ar + C = N), 64.40 (CH-N), 43.43 (CH2) ppm. FAB-MS: m/z = 455.9 [M]+.
2.3.3 Synthesis of L1-TMS
To an ice-cooled mixture of L1-Br (500 mg, 1.09 mmol) in freshly distilled triethylamine (20 mL) and CH2Cl2 (20 mL) solution under nitrogen was added Pd(OAc)2 (30 mg), PPh3 (90 mg) and CuI (30 mg). After the solution was stirred for 30 min, trimethylsilylacetylene (0.78 mL, 5.45 mmol) was then added and the suspension was stirred for another 30 min in the ice-bath before being warmed to room temperature. After reacting for 30 min at room temperature, the mixture was heated to 50 ℃ for 24 h. The solution was then allowed to cool to room temperature and the solvents were removed on a rotary evaporator in vacuo. The crude product was purified by column chromatography on silica gel eluting with hexane/CH2Cl2 (4 : 1, v/v) to provide L1-TMS (520 mg, 0.61 mmol, 57%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.64‒7.62 (m, 2 H, Ar), 7.47‒7.45 (m, 2 H, Ar), 7.31‒7.26 (m, 4 H, Ar), 7.26‒7.24 (m, 3 H, Ar), 6,96‒6.94 (m, 2 H, Ar), 5.35‒5.29 (m, 1H, CH), 3.85‒3.77 (m, 1H, CH2), 3.15‒3.10 (m, 1H, CH2), 0.26 (s, 9 H, Si(CH3)3), 0.21 (s, 9 H, Si(CH3)3) ppm. FAB-MS: m/z = 490.4 [M]+.
2.3.4 Synthesis of L1
A mixture of L1-TMS (163 mg, 0.33 mmol) and K2CO3 (114 mg, 0.85 mmol) in a mixture of methanol (5 mL) and CH2Cl2 (20 mL), under a nitrogen atmosphere, was stirred at room temperature overnight. The mixture was added to CH2Cl2 (30 mL) followed by washing with water (20 mL) three times and drying over anhydrous Na2SO4. After filtration, the solvents were removed on a rotary evaporator in vacuo. The crude product was purified by column chromatography on silica gel eluting with hexane/CH2Cl2 (6 : 1, v/v) to provide L1 (68 mg, 0.20 mmol, 61%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.68‒7.66 (m, 2 H, Ar), 7.51‒7.49 (m, 2 H, Ar), 7.34‒7.26 (m, 7 H, Ar), 7.00‒6.98 (m, 2 H, Ar), 5.37‒5.32 (m, 1H, CH), 3.89‒3.82 (m, 1H, CH2) 3.18‒3.12 (m, 2 H, CH2 + C ≡ CH), 2.97 (s, 1H, C ≡ CH) ppm. 13C NMR (100 MHz, CDCl3): δ = 146.83, 144.18, 141.65, 133.08, 132.68, 132.33, 129.31, 127.88, 125.68, 122.23, 113.02, 111.97 (Ar), 84.43, 83.52, 78.67, 75.48 (C ≡ C), 64.00 (Ph-C), 43.26 (CH2) ppm. FAB-MS: m/z = 346.1 [M]+. IR (KBr) (cm− 1): ν(C≡C−H) = 3275; ν(C≡C) = 2103.
2.3.5 Synthesis of P1
To a stirred mixture of L1 (34.6 mg, 0.10 mmol) and trans-[Pt(PBu3)2Cl2] (67.0 mg 0.10 mmol) in freshly distilled triethylamine (20 mL) and CH2Cl2 (20 mL) solution was added CuI (5 mg). The solution was stirred at room temperature for 24 h under a nitrogen atmosphere. The solvents were removed on a rotary evaporator in vacuo. The residue was redissolved in CH2Cl2 and filtered through a short aluminum oxide column using the same eluent to remove ionic impurities and catalyst residue. After removal of the solvent, the crude product was washed with hexane three times followed by methanol three times and then repeated precipitation from CH2Cl2/hexane (or CH2Cl2/methanol) and dried in vacuo to afford polymer P1 (68.0 mg, 71%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ = 7.55 (m, 2 H, Ar), 7.32‒7.26 (m, shielded by CHCl3 proton, 7 H, Ar), 7.08 (m, 2 H, Ar), 6.91‒6.88 (m, 2 H, Ar), 5.24 (m, 1H, CH), 3.83‒3.76 (m, 1H, CH2), 3.10‒3.08 (m, 1H, CH2), 2.13‒2.09 (m, 12 H, PC4H9), 1.60‒1.56 (m, 12 H, PC4H9), 1.45‒1.39 (m, 12 H, PC4H9), 0.94‒0.88 (m, 18 H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 3.04 (1JP−Pt = 2331 Hz) ppm. IR (KBr) (cm− 1): ν(C≡C) = 2096.
2.3.6 Synthesis of L2-Br
A mixture of L1-Br (675 mg, 1.49 mmol) and activated carbon (350 mg) was suspended in acetic acid (10 mL) and heated to 85 ℃ for 48 h under a nitrogen atmosphere. After the mixture was cooled down, water (50 mL) was added. The resulting solution was extracted with CH2Cl2 (30 mL) three times. The organic layer was combined and dried over anhydrous Na2SO4. The solvents were removed on a rotary evaporator in vacuo. The crude product was purified by column chromatography on silica gel eluting with hexane/CH2Cl2 (1:1, v/v) as eluent to provide L2-Br (410 mg, 0.90 mmol, 60%) as a white solid. 1H NMR (400 MHz, CDCl3): δ = 7.78 (d, J = 11.4 Hz, 2 H, Ar), 7.56 (d, J = 8.5 Hz, 2 H, Ar), 7.47 (d, J = 8.7 Hz, 2 H, Ar), 7.37‒7.35 (m, 3 H, Ar), 7.28‒7.26 (m, shielded by CHCl3 proton, 2 H, Ar), 7.24 (d, J = 8.7 Hz, 2 H, Ar), 6.79 (s, 1H, Ar) ppm. 13C NMR (100 MHz, CDCl3): δ = 151.14, 144.58, 138.93, 132.03, 131.79, 131.74, 130.02, 128.70, 128.67, 127.30, 126.50, 122.08, 121.09, 105.49 (Ar) ppm. FAB-MS: m/z = 455.1 [M]+.
2.3.7 Synthesis of L2-TMS
L2-TMS was prepared in a similar way as L1-TMS eluting with hexane/CH2Cl2 (1:1, v/v) to obtain the crude product which was a bit difficult to be purified but was used for the next step without further purification.
2.3.8 Synthesis of L2
Following the similar procedure as L1, L2 was obtained by direct desilylation of L2-TMS as a white solid in 60% using hexane/CH2Cl2 (1 : 1, v/v) as the eluent. 1H NMR (400 MHz, CDCl3): δ = 7.89‒7.87 (m, 2 H, Ar), 7.57‒7.55 (m, 2 H, Ar), 7.48‒7.46 (m, 2 H, Ar), 7.37‒7.32 (m, 5 H, Ar), 7.28‒7.26 (m, 2 H, Ar), 6.83 (s, 1H, Ar), 3.14 (s, 1H, C ≡ CH), 3.12 (s, 1H, C ≡ CH) ppm. 13C NMR (100 MHz, CDCl3): δ = 151.45, 144.63, 140.01, 133.18, 132.74, 132.50, 130.15, 128.74, 128.66, 125.59, 124.76, 121.61, 121.08, 105.89 (Ar), 83.68, 82.81, 78.24, 77.85 (C ≡ C) ppm. FAB-MS: m/z = 344.2 [M]+. IR (KBr) (cm− 1): ν(C≡C−H) = 3272; ν(C≡C) = 2107.
2.3.9 Synthesis of P2
Like P1, P2 was obtained as an off-white solid in 67% from L1. 1H NMR (400 MHz, CDCl3): δ = 7.78‒7.76 (m, 2 H, Ar), 7.31‒7.19 (m, shielded by the CHCl3 proton, 11 H, Ar) 6.78 (s, 1H, Ar), 2.14 (m, 12 H, PC4H9), 1.8 (m, 12 H, PC4H9), 1.46‒1.44 (m, 12 H, PC4H9), 0.94‒0.91 (m, 18 H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 3.01 (1JP−Pt = 2334 Hz) ppm. IR (KBr) (cm− 1): ν(C≡C) = 2098.
3 Results and Discussion
3.1 Synthesis and Characterization of Pt(II) Complexes
The chemical structure and synthetic routes of the polymers P1 and P2 are shown in Schemes 1 and 2, respectively. The key compound, L1-Br, was prepared by the ring closure reaction between (4-bromophenyl)hydrazine chloride and 1-(4-bromophenyl)-3-phenylprop-2-en-1-one in ethanol as a yellow solid. Ethanol was chosen as the solvent at the refluxing temperature to ensure the dissolution of the reactants and precipitation of the cyclized products. The use of acetic acid catalysts favours cyclization as opposed to the formation of phenylhydrazone derivatives of Michael addition products [27]. Following the general mechanism proposed by Coispeau and Elguero [28], 1,2-addition of the primary nitrogen atom of the phenylhydrazine occurs at the carbonyl group of ketone compound, followed by cyclization by means of an electrocyclic reaction involving six electrons. Conversion of the dibromide compound to its corresponding diethynyl congener (L1) via L1-TMS can be readily achieved following the typical organic synthetic protocols for alkynylation of aromatic halides followed by deprotection reaction using a base [29]. Polymer P1 was prepared by the Sonogashira-type dehydrohalogenation reaction between the diethynyl precursor (L1) and trans-[Pt(PBu3)2Cl2] with the feed mole ratio of 1:1 [3,4,5, 30]. It was carefully purified to remove ionic impurities and catalyst residues, and repeated precipitation and isolation led to an off-white solid with good yield and high purity.

The synthetic pathway of P1

The synthetic pathway of P2
Scheme 2 shows the chemical structure and synthetic strategy of P2. Polymer P2 was prepared from the dibromo precursor L2-Br using the same method as P1. Firstly, L2-Br was prepared from L1-Br as a white solid via the carbon-catalyzed oxidation reaction [31], which can then be ethynylated to give L2. Polymer P2 was similarly prepared as an off-white solid via the Sonogashira-type dehydrohalogenation reaction between the diethynyl precursor (L2) and trans-[Pt(PBu3)2Cl2].
The chemical structures of the Pt(II) complexes and polymers were verified by NMR (1H and 31P) and IR spectroscopy. The single 31P-{1H} NMR signal flanked by platinum satellites for each of the metallopolymers is consistent with a trans arrangement of the Pt(PBu3)2 unit in the square-planar geometry. The 1JP−Pt values of 2331–2334 Hz for the PBu3 moieties are typical of those for related trans-PtP2 systems [32, 33]. All the terminal alkynyl ligands and Pt(II) compounds display vibrational frequencies of the acetylenic functional groups clearly. In the IR spectra of the diethynyl ligands L1 and L2, they show IR νC≡C absorption at around 2103–2107 cm–1. The corresponding terminal acetylenic C ≡ C–H stretching vibrations occur at around 3272–3275 cm–1. The C ≡ C stretching frequencies for P1–P2 are lower than those for L1-L2, in line with a higher degree of conjugation in the polymers [34].
Using gel-permeation chromatography (GPC), the number-average molecular weights (Mn) of P1 and P2 were estimated to be 28,890 and 30,075, respectively, calibrated against polystyrene standards (Table 1). Their polydispersity indexes (PDI) in the narrow range of 1.24–1.49 are consistent with the proposed linear structure from the condensation polymerization [3]. It is known that GPC does not give absolute molecular weights but provides a measure of hydrodynamic volume. We expect a notable difference in the hydrodynamic behavior between rigid-rod type polymers in solution and flexible polymers. So, these molecular weights may be overestimated, leading to certain systematic errors in the GPC data.
3.2 Thermal, Optical and Redox Properties
Both polymers are thermally and air-stable solids and they are soluble in common organic solvents. From the curves in thermal gravimetric analysis (TGA), the respective percent weight loss indicates the removal of one PBu3 and some of the Bu groups from their polymers in the decomposition step. Decomposition onset was defined by a 5 wt.-% loss in each case. The onset decomposition temperatures of 322 °C (P1) and 348 °C (P2) are notably higher than those for the polymers with diphenyl (297 °C) [35] and dipyridyl (274 °C) [36] moieties.
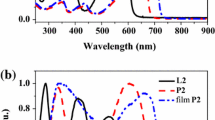
The polymers P1 and P2 exhibit different absorption properties. Figure 1 shows the UV/Vis absorption of L1 and P1 and PL emission spectra of P1 at 293 K and 77 K. Those of L2 and P2 are depicted in Fig. 2. The absorption spectrum of L1 is characterized by a broad and featureless band peaking at 391 nm which can be assigned to a ligand-centered π-π* transition. The other one below 300 nm is the characteristic absorption peak of the triaryl-pyrazoline group. However, L2 only shows one peak below 300 nm, which is due to the pyrazolyl group. The absorption bands of P1 and P2 have a significant red shift compared to those of the corresponding ligands, which can be attributed to the extension of metal-organic π-conjugation length in the presence of heavy platinum unit.

Normalized absorption and emission spectra of L1 and P1 in CH2Cl2 solution at 293 K and 77 K

Normalized absorption and emission spectra of L2 and P2 in CH2Cl2 solution at 293 K and 77 K
P1 and P2 show similar broad and structureless emission peaks at around 514 nm arising from the singlet excited state at 293 K. The PL quantum yields of P1 and P2 in CH2Cl2 were measured to be 3.2 and 2.8%. The measured PL lifetimes for P1 and P2 for the main peaks are all very short (ca. 0.58–0.66 ns) at 293 K, characteristic of the spin-allowed singlet emission [1, 3]. With reference to other pyrazole derivatives in the literature, since this emission peak is observed with a significant Stokes shift, the absorption transition probably consists of π-π* origin with some charge transfer character to establish a lower-energy twisted intramolecular charge-transfer state [37]. A strong π-π* triplet emission at around 494 and 506 nm, with a triplet emissive lifetime τP of ca. 69.9 µs and 78.9 µs, is detectable from the PL spectrum of P1 and P2 at 77 K, respectively. The long lifetime is caused by the heavy metal center which induces strong spin-orbit coupling, and then increases the phosphorescence [1, 3]. The small peak at 401 nm for P1 is due to the weak π-π* fluorescence emission, typical of other Pt-polyynes reported in the literature [38, 39].
Both polymers undergo a rigidochromic blue shift in the emission wavelength upon cooling of their solutions to 77 K. The hypsochromic shift observed at low temperature is mainly caused by the solvent reorganization in a fluid solution at 77 K that can readily stabilize the charge transfer states prior to emission [40]. This process is significantly impeded in a rigid matrix at 77 K, and hence fluorescence appears at a higher energy. The absence of vibronic progression in the emission profile (at 293 K) suggests mostly a charge-transfer state but not the ligand-centered π-π* excited state. The observed emission lifetimes (τ) of our platinum compounds at 77 K are longer than those measured at room temperature [41] (Table 2).
The HOMO and LUMO levels of the polymers P1–P2 were calculated based on the redox potentials determined from electrochemical measurements using cyclic voltammetry. The key data are gathered in Table 3. From the onset values of oxidation potential (Eonset, ox), the HOMO levels of the polymers were calculated according to the equation EHOMO = -(Eonset, ox + 4.72) eV where the unit of potential is V versus Ag/AgCl while the LUMO levels were determined from the optical bandgap and the energy level of HOMO using the equation ELUMO = (EHOMO + Egopt) eV [42, 43]. P1 shows an irreversible oxidation wave at 0.64 eV while P2 shows an oxidation wave at 1.12 eV. P1 shows an elevated HOMO level (-5.36 eV) relative to P2 (-5.84 eV), indicating that P1 is more electropositive (or has a lower ionization potential) than P2, and hence a better hole transport ability in P1 would be anticipated.
3.3 Photovoltaic Properties
Since the light-induced intramolecular electron transfer could easily occur from donor to acceptor through the π-conjugation which favours photocurrent generation and the photoelectronic energy conversion in photovoltaic devices, PSCs with BHJ configuration were fabricated by using P1 with better absorption properties as electron donor and PCBM as electron acceptor. The hole collection electrode consisted of indium tin oxide (ITO) with a spin-coated poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS), while Al served as the electron collecting electrode. Figure 3 shows the J-V curve of PSCs with P1:PCBM (1:4, w/w) active layers under simulated AM1.5 solar irradiation. A PCE of 0.27% can be obtained for P1 with Voc = 0.63 V, Jsc = 1.33 mA cm− 2 and FF = 0.32 under illumination of an AM 1.5 solar cell simulator.

J-V curve of PSC with P1:PCBM (1:4 w/w) active layer under simulated AM 1.5 solar irradiation
4 Conclusion
In summary, two new platinum-containing polymetallayne polymers functionalized with pyrazoline and electron-rich pyrazole moieties have been successfully synthesized via Sonogashira-type dehydrohalogenation polycondensation reaction. All of them exhibit good solubility in common organic solvents and good thermal stability and show different absorption properties in the visible region. This observation showcases that the energy level and bandgap of the polymers could be effectively tuned by changing the number of double bonds of the five-membered ring of the pyrazoline derivative. It is found that the use of pyrazole down-shifted the HOMO energy level by ca. 0.48 eV and blue-shifted the absorption profile of the conjugated polymer as compared to the corresponding pyrazoline counterpart. These results reveal that molecular control by using pyrazoline instead of pyrazole ring would broaden the absorption and up-shifting the HOMO level, hence improving the photovoltaic property of PSCs.
Data Availability
No datasets were generated or analysed during the current study.
References
A. Haque, R.A. Al-Balushi, I.J. Al-Busaidi, M.S. Khan, P.R. Raithby, Chem. Rev. 118, 8474–8597 (2018)
P.D. Harvey, J. Inorg. Organomet. Polym. Mater. 27, S3–S38 (2017)
C.-L. Ho, Z.-Q. Yu, W.-Y. Wong, Chem. Soc. Rev. 45, 5264–5295 (2016)
C.-L. Ho, W.-Y. Wong, Coord. Chem. Rev. 257, 1614–1649 (2013)
W.-Y. Wong, C.-L. Ho, Coord. Chem. Rev. 250, 2627–2690 (2006)
A. Haque, L. Xu, R.A. Al-Balushi, M.K. Al-Suti, R. Ilmi, Z. Guo, M.S. Khan, W.-Y. Wong, P.R. Raithby, Chem. Soc. Rev. 48, 5547–5563 (2019)
W.-Y. Wong, Dalton Trans. 4495–4510 (2007)
W.-Y. Wong, P.D. Harvey, Macromol. Rapid Commun. 31, 671–713 (2010)
W.-Y. Wong, C.-L. Ho, Acc. Chem. Res. 43, 1246–1256 (2010)
A. Haque, R.A. Al-Balushi, M.S. Khan, J. Organomet. Chem. 897, 95–106 (2019)
G.-J. Zhou, W.-Y. Wong, Chem. Soc. Rev. 40, 2541–2566 (2011)
W.-Y. Wong, J. Inorg. Organomet. Polym. Mater. 15, 197–219 (2005)
J. Barberá, K. Clays, R. Giménez, S. Houbrechts, A. Persoons, J.L. Serrano, J. Mater. Chem. 8, 1725–1730 (1998)
M. Mehkoom, S.M. Afzal, S. Ahmad, S.A. Khan, J. Mol. Liq. 345, 117018 (2022)
M. Khalid, A. Mustafa, S. Ahmed, M.A. Asghar, T. Ahamad, A.A. Braga, S.C. Ojha, Arab. J. Chem. 16, 105271 (2023)
M. Parshad, D. Kumar, V. Verma, Inorg. Chim. Acta. 560, 121789 (2024)
J.O. Morley, D. Pugh, in Organic Materials for Nonlinear Optics, ed. R. A. Hann and D. Bloor, Spec. Publ. R. Soc. Chem. 69, Kent, 1989, p. 28
E. David, K. Mishra, K. Thirumoorthy, N. Palanisami, Appl. Organomet. Chem. 35, e6138 (2021)
Y.S. Xie, X.-H. Pan, B.-X. Zhao, J.-T. Liu, D.-S. Shin, J.-H. Zhang, L.-W. Zheng, J. Zhao, J.-Y. Miao, J. Organomet. Chem. 693, 1367–1374 (2008)
K. Senthilkumar, K. Thirumoorthy, G. Vinitha, K. Soni, N.S.P. Bhuvanesh, N. Palanisami, J. Mol. Struct. 1128, 36–43 (2017)
M. Pokladko-Kowar, E. Gondek, A. Danel, T. Uchacz, P. Szlachcic, K. Wojtasik, P. Karasinski, Crystals. 12, 434 (2022)
C.-Q. Ma, L.-Q. Zhang, X.-H. Li, X.-S. Wang, B.-W. Zhang, Y. Cao, D.-M. Wang, X.-Y. Jiang, Z.-L. Zhang, D.-Q. Zhang, Y. Qiu, Acta Chim. Sinica. 60, 847–853 (2002)
C.-Q. Ma, L.-Q. Zhang, J.-H. Zhou, X.-S. Wang, B.-W. Zhang, Y. Cao, P. Bugnon, M. Schaer, F. Nüesch, D.-Q. Zhanga, Y. Qiu, Chin. J. Chem. 20, 929–932 (2002)
D. Xiao, L. Xi, W. Yang, H. Fu, Z. Shuai, Y. Fang, J. Yao, J. Am. Chem. Soc. 125, 6740–6475 (2003)
V. Mukundam, S. Sa, A. Kumari, T.T. Ponduru, R. Das, K. Venkatasubbaiah, Chem. Asian J. 17, e202200291 (2022)
G.B. Kauffman, L.A. Teter, Inorg. Synth. 7, 245–249 (1963)
R.S. Theobald, in Rodd’s Chemistry of Carbon Compounds, 2nd Edition, ed. S. Coffey and M. F. Ansell, Vol. 4, Heterocyclic Compounds, Part C, ed. M. F. Ansell, Elsevier, New York, 1986, ch. 1
G. Coispeau, J. Elguero, Bull. Soc. Chim. Fr. 2717–2736 (1970)
S. Takahashi, Y. Kuroyama, K. Sonogashira, N. Hagihara, Synthesis 627–630 (1980)
W.-Y. Wong, C.K. Wong, G.L. Lu, A.W.M. Lee, K.W. Cheah, J.X. Shi, Macromolecules. 36, 983–990 (2003)
N. Nakamichi, Y. Kawashita, M. Hayashi, Synthesis. 7, 1015–1020 (2004)
W.-Y. Wong, C.K. Wong, G.L. Lu, K.W. Cheah, J.X. Shi, Z. Lin, J. Chem. Soc. Dalton Trans. 4587–4594 (2002)
Q. Wang, W.-Y. Wong, Polym. Chem. 2, 432–440 (2011)
J. Lewis, N.J. Long, P.R. Raithby, G.P. Shields, W.-Y. Wong, M. Younus, J. Chem. Soc. Dalton Trans. 4283–4288 (1997)
L. Liu, S.-Y. Poon, W.-Y. Wong, J. Organomet. Chem. 690, 5036–5048 (2005)
M.S. Khan, M.R.A. Al-Mandhary, M.K. Al-Suti, A.K. Hisahm, P.R. Raithby, B. Ahrens, M.F. Mahon, L. Male, E.A. Marseglia, E. Tedesco, R.H. Friend, A. Köhler, N. Feeder, S.J. Teat, J. Chem. Soc. Dalton Trans. 1358–1368 (2002)
S.K. Liew, A. Holownia, A.J. Tilley, E.I. Carrera, D.S. Seferos, A.K. Yudin, J. Org. Chem. 81, 10444–10453 (2016)
W.-Y. Wong, G.-L. Lu, K.-H. Choi, J.-X. Shi, Macromolecules. 25, 3506–3513 (2002)
N. Chawdhury, A. Köhler, R.H. Friend, W.-Y. Wong, J. Lewis, M. Younus, P.R. Raithby, T.C. Corcoran, M.R.A. Al-Mandhary, M.S. Khan, J. Chem. Phys. 110, 4963–4970 (1999)
A.B. Tamayo, S. Garon, T. Sajoto, P.I. Djurovich, I.M. Tsyba, R. Bau, M.E. Thompson, Inorg. Chem. 44, 8723–8732 (2005)
C.-L. Ho, W.-Y. Wong, G.-J. Zhou, B. Yao, Z. Xie, L. Wang, Adv. Funct. Mater. 17, 2925–2936 (2007)
J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang, Y. Li, J. Am. Chem. Soc. 128, 4911–4916 (2006)
C.G. Van de Walle, J. Neugebauer, Nature. 423, 626–628 (2003)
Acknowledgements
W.-Y. W. thanks the financial support from the RGC Senior Research Fellowship Scheme (SRFS2021-5S01), the National Natural Science Foundation of China (52073242), the Hong Kong Research Grants Council (PolyU 15307321), Research Institute for Smart Energy (CDAQ) and Miss Clarea Au for the Endowed Professorship in Energy (847 S). Q. W. and J. L. thank the financial support from Chengdu University. Z. S. thanks the financial support from the Hong Kong Polytechnic University (1-BDTU).
Funding
Open access funding provided by The Hong Kong Polytechnic University
Author information
Authors and Affiliations
Contributions
Qiwei Wang: conceptualization, investigation, resources, supervision, writing-original draft; Lu Jiang: data curation, investigation; Junlong Li: data curation, funding acquisition; Zelin Sun: funding acquisition, writing-review & editing; Wai-Yeung Wong: funding acquisition, project administration, resources, supervision, writing-review & editing.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Q., Jiang, L., Li, J. et al. Polyplatinaynes Functionalized with Pyrazoline Derivatives. J Inorg Organomet Polym (2024). https://doi.org/10.1007/s10904-024-03125-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10904-024-03125-0