
Abstract
Four new solution-processable platinum-containing polyynes functionalized with triphenylamine backbone and different acceptor fragment in the side chain were synthesized and characterized by spectroscopic, thermal and optical methods. The main- and side-chains show different absorption features in the solar spectrum, resulting in broad absorption coverage of the whole visible region. By changing the acceptor group in the side chain, the photophysical properties including energy levels, absorption wavelength and bandgap of the polymers were finely tuned. As the strength of electron acceptor is increased, the power conversion efficiency (PCE) of organic polymer solar cells fabricated with these polymers as electron donor and (6,6)-phenyl-C61-butyric acid methyl ester (PCBM) as electron acceptor was increased. PCE of 1.35% can be obtained from solar cell based on the polyplatinayne with the dicyanovinyl side group under illumination of an AM 1.5 solar cell simulator in a 1:4 (polymer:PCBM) blend ratio.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Organic polymer solar cells (PSCs) have attracted considerable attention because of their unique advantages, such as low fabrication cost, light-weight, flexibility as well as large area of device and have great potential to be used as flexible devices [1,2,3,4,5,6,7,8,9]. Bulk heterojunction (BHJ) configuration of PSCs has received extensive studies, because such configuration creates large interfacial areas between the polymers and electron acceptors (e.g. fullerene derivatives, (6,6)-phenyl-C61-butyric acid methyl ester (PCBM)), thus leading to efficient photoinduced charge separation in the device [1, 2, 10,11,12,13]. In this context, low-bandgap conjugated polymers have shown great promises for photoinduced charge generation in BHJ cells to capture a large portion of solar energy. Previously, the power conversion efficiency (PCE) of PSCs based on poly(3-hexylthiophene-2,5-diyl) (P3HT) thin film blended with PCBM has reached up to 4–5% [2, 14, 15]. However, to further improve the efficiency of P3HT-based PSCs its absorption must be broadened in order to match well with the solar spectrum. Since approximately 54% of the sunlight coverage is distributed from 380 to 800 nm, an ideal active layer for a PSC should have a broad and strong absorption spectrum in this range. The mismatch between the absorption spectrum of the photoactive layer and the sunlight spectrum leads to low short-circuit current density (Jsc), which is responsible for the low PCE of the PSCs in the past. Later on, PSCs fabricated with low bandgap polymers with high Jsc reached a PCE over 5% [16,17,18,19,20]. A key characteristic of conjugated polymers is the easy modification of their molecular structure and properties by side chain substitution or copolymerization. The substituents added in the side chain of the polymer could result in the lowering of their bandgaps. Moreover, the structure of these polymers results in the improvement of the isotropic charge transport, which is important for highly efficient PSCs [21,22,23,24,25,26,27]. Besides, triarylamine derivatives have propeller shape and strong electron-donating ability, which have been widely used as an electron donor in photoactive materials to afford promising PCEs with improved hole mobility of their PSCs [28,29,30,31,32,33,34].
As an alternative class of conjugated polymers, it would be interesting to integrate metallated chromophore into the conjugated polymer structures [35,36,37,38]. The introduction of metal ions into conjugated polymers offers various possible merits [35,36,37,38]: (a) the metal ions may act as architectural templates in the assembly of the organic subunits; (b) they may provide redox-active and paramagnetic centers to generate active species for charge transport and may alter the electronic and optical properties of organic π-systems; (3) they allow fine-tuning of the HOMO–LUMO gap through the interaction of the metal d-orbitals with the ligand orbitals; (4) there is a diversity of molecular frameworks based on different coordination number, geometry and valence shells of different metal atoms. Among these, platinum-containing polymers are good candidates for PSCs since they have high charge-carrier mobility and good absorption properties [39,40,41]. In this work, we designed a series of new polyplatinayne polymers, which contain both main-chain and side-chain chromophores. The absorption of these polymers was expected to match a majority of the visible part of the sunlight spectrum. Figure 1 shows the design and general structure of the polymers. The main chain and side chain were connected by the Z unit, such as nitrogen atom. The main chain and the side chain would exhibit different absorption range in the solar spectrum. Herein, we report the synthesis and characterization of four new polymetallayne polymers of Pt with triphenylamine backbone and different electron acceptors in the side chain. Photovoltaic properties of these polymers were investigated by fabricating BHJ photovoltaic devices using these polymers as the electron donor and PCBM as the electron acceptor. The results indicated that the Jsc and PCE of their PSCs can be significantly enhanced by increasing the strength of the acceptor in the side chain. BHJ solar cells fabricated from these polymers gave the best PCE of 1.35% with a Jsc of 4.47 mA cm−2 under illumination of an AM 1.5 solar cell simulator.

The general skeleton of the polyplatinaynes containing both main chain and side chain chromophores
2 Experimental Section
2.1 Materials and Instruments
All the chemicals were purchased from Acros or Aldrich and used as received unless otherwise specified. 5′-Bromo-2,2′-bithiophene-5-carbaldehyde [42,43,44], trans-[Pt(PEt3)2PhCl] [45] and trans-[Pt(PBu3)2Cl2] [46] were prepared using the methods reported in the literature. THF was dried by distillation from sodium with benzophenone as an indicator under a nitrogen atmosphere. Separation and purification of products were achieved by column chromatography on silica gel. TLC was carried out in air using laboratory grade solvents as eluents.
The positive-ion fast atom bombardment (FAB) mass spectra were recorded in m-nitrobenzyl alcohol matrices on a Finngin-MAT SSQ710 mass spectrometer. Infrared spectra were recorded on the Nicolet Magna 550 Series II FTIR spectrometer, using KBr pellets for solid state spectroscopy. NMR spectra were measured in deuterated solvents as the lock and reference on Varian INOVA 400 instrument or Bruker AV 400 MHz FT-NMR spectrometer, with 1H and 13C NMR chemical shifts quoted relative to Me4Si standard and 31P chemical shifts relative to an 85% H3PO4 external reference. Electronic absorption spectra were obtained with a Hewlett Packard 8453 spectrometer. Solution state photoluminescence measurements were obtained by the LS50B fluorescent spectrometer. For lifetime measurements, the third harmonics, 355 nm line of a Q-switched Nd:YAG laser was used as the excitation light source. The emission was recorded by using a PMT and a HP54522A 500 MHz oscilloscope. The PL spectra were measured in CH2Cl2 with a PTI Fluorescence Master Series QM1 spectrophotometer. The molecular weights of the polymers were determined by gel permeation chromatography (GPC) (HP 1050 series HPLC with visible wavelength and fluorescent detectors) using polystyrene standards. Thermal analysis was performed with a Perkin-Elmer TGA6 thermal analyzer.
2.2 Fabrication and Characterization of Polymer Solar Cells
PCBM was purchased from American Dyes while PEDOT/PSS (Baytron P VPAI 4083) was purchased from H. C. Starck. Device configuration of ITO/poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS)/polymer:PCBM/Al was applied in this study. Indium tin oxide (ITO) coated glass substrates (10 Ω per square) were cleaned by sonication in toluene, acetone, ethanol, and deionized water, dried in an oven, and then cleaned with UV ozone for 300 s. As-received PEDOT:PSS solution was passed through the 0.45 μm filter and spin-coated on patterned ITO substrates at 5000 rpm for 3 min, followed by baking in N2 at 150 °C for 15 min. P1–P4:PCBM (1:4 by weight) active layer was prepared by spin-coating the toluene solution (4 mg mL–1 of metallopolyyne and 16 mg mL–1 of PCBM) at 800 rpm for 2 min. The substrates were dried at room temperature under vacuum for 1 h and then stored under high vacuum (10–5–10–6 Torr) overnight. An Al electrode (100 nm) was evaporated through a shadow mask to define the active area of the devices (2 mm2 circle). All the fabrication procedures (except drying, PEDOT:PSS annealing, and Al deposition) and cell characterization were performed in air. Power conversion efficiency (PCE) was determined from J–V curve measurement (using a Keithley 2400 sourcemeter) under white light illumination (at 100 mW cm–1). For white light efficiency measurements, an Oriel 66002 solar light simulator with an AM1.5 filter was used. The light intensity was measured by a Molectron Power Max 500D laser power meter. For the measurement of the external quantum efficiency (EQE), different wavelengths were selected with an Oriel Cornerstone 74000 monochromator, while the photocurrent was measured with a Keithley 2400 sourcemeter. The light intensity was measured with a Newport 1830-C optical power meter equipped with a 818-UV detector probe.
2.3 General Procedures for the Synthesis of Polyplatinayne Polymers P1–P2
The synthesis of P1 was taken as a typical example.
To a stirred mixture of L1 and trans-[Pt(PBu3)2Cl2] in a 1:1 molar ratio in freshly distilled triethylamine and CH2Cl2 solution (1:1, v/v), CuI (3.0 mg) was added. The solution was stirred at room temperature for 24 h under a nitrogen atmosphere. The solvent was removed on a rotary evaporator in vacuo. The residue was dissolved in CH2Cl2 and filtered through a short aluminum oxide column using the same eluent to remove ionic impurities and catalyst residue. After removal of the solvent, the crude product was washed with hexane followed by methanol and then repeated precipitation from CH2Cl2/hexane and dried in vacuo to afford polymer P1 (62%) as a red solid. 1H NMR (400 MHz, CDCl3): δ = 9.85 (s, 1H, CHO), 7.68 (m, 1H, Ar), 7.45–7.42 (m, 2H, Ar), 7.32 (m, 1H, Ar), 7.24 (m, 1H, Ar), 7.18–7.16 (m, 5H, Ar), 7.0–7.03 (m, 2H, Ar), 6.97–6.94 (m, 4H, Ar), 2.17–2.13 (m, 12H, PC4H9), 1.62–1.56 (m, 12H, PC4H9), 1.48–1.43 (m, 12H, PC4H9), 0.93 (t, J = 7.2 Hz, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 2.76 (1JP-Pt = 2345 Hz) ppm. IR (KBr) (cm−1): 2098 (νC≡C); 1667 (νCHO).
P2: Red solid. Yield: 65%. 1H NMR (400 MHz, CDCl3): δ = 9.86 (s, 1H, CHO), 7.68 (m, 1H, Ar), 7.50–7.44 (m, 6H, Ar), 7.33 (m, 1H, Ar), 7.26 (m, 1H, Ar), 7.20 (m, 1H, Ar), 7.13–7.09 (m, 6H, Ar), 7.04 (m, 2H, Ar), 6.80 (m, 2H, Ar), 2.15–2.11 (m, 12H, PC4H9), 1.62–1.58 (m, 12H, PC4H9), 1.51–1.46 (m, 12H, PC4H9), 0.95 (t, J = 7.2 Hz, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 3.29 (1JP-Pt = 2315 Hz) ppm. IR (KBr) (cm–1): 2085 (νC≡C); 1666 (νCHO).
2.4 General Procedures for the Synthesis of Polyplatinayne Polyynes P3–P4
The synthesis of P3 was taken as a typical example.
To a mixture of P1 (40 mg) and malononitrile (3 mg) in dry chloroform (10 mL), triethylamine (1 drop) was added under a nitrogen atmosphere. The resulting mixture was heated to reflux for 2 h. After cooling down to room temperature, the solvents were removed and methanol (20 mL) was added. The precipitate was collected and washed with methanol and dried to provide P3 (38 mg, 95%) as a dark red solid. 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H, HC = C(CN)2), 7.63 (m, 2H, Ar), 7.45–7.40 (m, 3H, Ar), 7.26 (m, shielded by CHCl3 proton, 1H, Ar) 7.21–7.17 (m, 5H, Ar), 7.05–7.03 (m 2H, Ar), 6.97–6.95 (m, 4H, Ar), 2.15–2.13 (m, 12H, PC4H9), 1.60–1.59 (m, 12H, PC4H9), 1.48–1.43 (m, 12H, PC4H9), 0.95–0.88 (m, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 2.78 (1JP-Pt = 2348 Hz) ppm. IR (KBr) (cm–1): 2220 (νCN); 2095 (νC≡C).
P4: Dark red solid. Yield: 89%. 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H, C = CH), 7.63 (m, 1H, Ar), 7.50–7.47 (m, 6H, Ar), 7.41 (m, 1H, Ar), 7.27 (m, 1H, Ar), 7.13–7.09 (m, 6H, Ar), 7.05 (m, 2H, Ar), 7.80 (m, 2H, Ar), 2.15–2.11 (m, 12H, PC4H9), 1.62–1.56 (m, 12H, PC4H9), 1.51–1.46 (m, 12H, PC4H9), 0.97–0.94 (m, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 3.30 (1JP-Pt = 2318 Hz) ppm. IR (KBr) (cm–1): 2221 (νCN); 2084 (νC≡C).
2.5 General Procedures for the Synthesis of Dinuclear Pt(II) Model Complexes M1–M2
The synthesis of M1 was taken as a typical example.
To a stirred mixture of L1 (24 mg, 0.03 mmol) and trans-[Pt(PEt3)2PhCl] (35 mg, 0.064 mmol) in freshly distilled triethylamine (10 mL) and CH2Cl2 (10 mL) was added CuI (3.0 mg). The solution was stirred at room temperature under nitrogen over a period of 24 h. After removal of the solvent, the crude product was purified by column chromatography on silica gel eluting with hexane/CH2Cl2 (1:1, v/v) to give M1 (57%) as a red solid. 1H NMR (400 MHz, CDCl3): δ = 9.85 (s, 1H, CHO), 7.67 (d, J = 4.0 Hz, 1H, Ar), 7.43 (d, J = 8.8 Hz, 2H, Ar), 7.34–7.31 (m, 5H, Ar), 7.24 (d, J = 4.0 Hz, 1H, Ar), 7.20 (d, J = 8.4 Hz, 4H, Ar), 7.16 (d, J = 3.6 Hz, 1H, Ar), 7.05 (d, J = 8.8 Hz, 2H, Ar), 6.98–6.94 (m, 8H, Ar), 6.82–6.78 (m, 2H, Ar), 1.80–1.74 (m, 24H, PC2H5), 1.14–1.06 (m, 36H, PC2H5) ppm. 13C NMR (100 MHz, CDCl3): δ = 182.41 (CHO), 156.42, 148.04, 147.60, 146.59, 143.67, 141.12, 139.20, 137.51, 135.21, 133.73, 131.80, 127.28, 126.45, 126.28, 125.02, 124.60, 124.54, 123.64, 122.89, 122.37, 121.19 (Ar), 112.75, 109.68 (C≡C), 15.24, 15.08, 14.90, 8.05 (PC2H5) ppm. 31P NMR (161 MHz, CDCl3): δ = 9.87 (1JPt-P = 2625 Hz) ppm. FAB-MS (m/z): 1500.7 [M + 1]+. IR (KBr) (cm–1): 2093 (νC≡C); 1664 (νCHO).
M2: Red solid. Yield: 44%. 1H NMR (400 MHz, CDCl3): δ = 9.86 (s, 1H, CHO), 7.67 (d, J = 4.0 Hz, 1H, Ar), 7.49–7.45 (m, 6H, Ar), 7.33–7.31 (m, 5H, Ar), 7.25 (d, J = 4.0 Hz, 2H, Ar), 7.20 (d, J = 4.0 Hz, 1H, Ar), 7.14–7.08 (m, 6H, Ar), 7.04 (d, J = 3.6 Hz, 2H, Ar), 6.99–6.95 (m, 4H, Ar), 6.83–6.81 (m, 4H, Ar), 1.78–1.72 (m, 24H, PC2H5), 1.14–1.06 (m, 36H, PC2H5) ppm. 13C NMR (100 MHz, CDCl3): δ = 182.43 (CHO), 155.92, 147.45, 147.43, 146.17, 145.60, 141.27, 139.86, 139.07, 138.98, 137.48, 134.15, 130.16, 129.48, 128.02, 127.43, 127.35, 127.27, 126.67, 126.33, 124.78, 123.78, 123.47, 123.29, 122.16 (Ar), 121.35, 102.41 (C≡C), 15.30. 15.13, 14.96, 8.05 (PC2H5) ppm. 31P NMR (161 MHz, CDCl3): δ = 9.96 (1JPt-P = 2613 Hz) ppm. FAB-MS (m/z): 1663.6 [M + 1]+. IR (KBr) (cm–1): 2080 (νC≡C); 1663 (νCHO).
3 Results and Discussion
3.1 Synthesis and Characterization of Pt(II) Complexes
The synthetic routes of dinuclear model compounds M1–M2 and polymers P1–P4 are shown in Schemes 1 and 2, respectively. The experimental details of the diethynyl precursors are described in the supporting information [47]. The polymers P1–P2 were prepared by the Sonogashira-type dehydrohalogenation reaction between platinum dichloride precursors and the corresponding diethynyl ligands [48,49,50,51,52]. The feed mole ratio of the platinum precursors and the ethynyl ligands were 1:1 and 2:1 for the synthesis of polymers P1–P2 and dimers M1–M2, respectively, and each product was purified carefully to remove ionic impurities and catalyst residues. The dinuclear Pt complexes serve as good discrete molecular model complexes for the corresponding polymers as far as their spectroscopic and photophysical properties are concerned. However, to our surprise, for the polymers P3 and P4, they cannot be prepared from their diethynyl precursors, which may be due to the poor stabilities of their respective precursors under the reaction condition. Instead, they were successfully synthesized from their respective polymers P1 and P2 in high yields via the Knoevenagel condensation reaction using the facile procedure of treating P1 or P2 with malononitrile in chloroform in the presence of a catalytic amount of triethylamine [44, 53]. The polymers P3 and P4 can be purified by column chromatography over aluminium oxide followed by the repeated precipitation and isolated in good yields and high purity. All of these Pt(II) compounds are thermally and air-stable solids and they are soluble in common chlorinated hydrocarbons and toluene.

Synthetic routes to dinuclear model compounds M1–M2

Synthetic routes to polymers P1–P4
The chemical structures of the Pt(II) complexes and polymers were verified by NMR (1H and 31P) and IR spectroscopies. The single 31P-{1H} NMR signal flanked by platinum satellites for each of the platinum(II) complexes is consistent with a trans geometry of the Pt(PBu3)2 or Pt(PEt3)2 in such square-planar geometry. The 1JP−Pt values of around 2320 Hz for the PBu3 moieties and 2620 Hz for the PEt3 are typical of those for related trans-PtP2 systems [48,49,50,51,52]. All the terminal alkynyl ligands and Pt(II) compounds were subjected to IR measurements in which the vibrational frequencies of the acetylenic functional groups were detected. In the IR spectra of the diethynyl ligands, L1–L2 display IR νC≡C absorption at around 2095‒2102 cm–1. The corresponding terminal acetylenic C≡C–H stretching vibrations occur at around 3271‒3277 cm–1. The C≡C stretching frequencies for P1–P4 are located in the range of 2084–2098 cm–1, which are lower than those for the corresponding free alkynyl precursors, in line with a higher degree of conjugation in the former [50]. The νCHO vibrations appear at the region of 1663‒1667 cm–1 for M1‒M2 and P1‒P2 whereas the νC≡N stretching bands vibrate at 2220 cm‒1 for P3–P4.
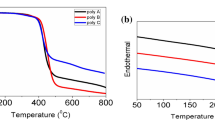
From the results of gel-permeation chromatography (GPC), the number-average molecular weights (Mn) of P1–P4 were calibrated against polystyrene standards, which ranged from 12,130 to 18,330 g/mol (Table 1). Their polydispersity indexes (PDI ~ 1.80–1.93) are consistent with the proposed linear structure from the condensation polymerization [48,49,50,51,52]. The thermal properties of the polymers were also examined by thermal gravimetric analysis (TGA) under a nitrogen atmosphere (Table 1). All the polymers exhibit good thermal stability with the decomposition onsets over 330 °C.
3.2 Optical Properties
The photophysical properties of all new Pt(II) complexes were investigated by UV-Vis and photoluminescence (PL) spectroscopies in CH2Cl2 solution and the related data are summarized in Table 2. All the compounds were characterized by two bands in their absorption spectra (Fig. 2). The first intense absorption band in the range of 270–450 nm is mainly attributed to the π-π* transition in the metal–organic π-conjugation system. The second one appeared from 450 nm and up to 650 nm is assigned to the intramolecular charge transfer (ICT) band between the π-conjugated main chain and the acceptor unit in the side chain [54]. Because of such ICT interaction, there is a notable narrowing of the bandgap (Eg) from the value of 2.72 eV for the Pt-polymer with a methyl substituent on the triphenylamine moiety [55] to the value of 2.30 eV for P2 and 1.91 eV for P4. Relative to the free alkynes (λmax = 346, 421 nm for L1; 398 nm for L2, see Figs. S3 and S4), there is a slight red shift in the absorption wavelength for their corresponding platinum(II) compounds. The spectroscopic properties of these complexes offer a quick guide to their effective conjugation length and bandgaps. Interestingly, the addition of an extra thiophene ring in the main chain in P2 does not affect much the value of Eg as compared to P1. Instead, the π-π* absorption of P2 is red-shifted relative to P1, indicating that P2 has a slightly longer effective π-conjugation. Moreover, due to the bathochromic shift of the π-π* bands in P2, overlapping of its π-π* and ICT absorption bands was detected. Similar phenomena were observed in their corresponding dinuclear complexes M1–M2. The molar extinction coefficients (ε) can also be enhanced by the addition of peripheral thienyl group in P1–P2. The triphenylamine at the core of polymers P1–P2 connects the conjugated moieties via three-way branched network, which serves as a spacer and a conjugated interrupter, thus effectively controlling the conjugated length of the polymers (Fig. 3). Therefore, the λmax between polymers and their corresponding diynes do not show a significant red shift. Taking advantage of the strongly electron-accepting dicyanovinyl functional group at the side chain, λmax with 536 and 528 nm were measured in P3 and P4, respectively, which display a broader absorption coverage in comparison to P1 and P2. Compared to P1 and P2, the second band of P3 and P4 is red-shifted by about 80 nm due to the stronger ICT interaction in the presence of the strong electron-accepting CN groups in the side chain [56]. Hence, the Eg value is lowered from P1 and P2 to P3 and P4 by replacing the aldehyde group with the dicyanovinyl moiety. It is worth noting that, apart from the conjugated main chain backbones as the efficient light-harvesting chromophores, functionalized side chain would also serve as the effective absorption antennae.

Normalized absorption spectra of M1–M2 and P1–P4 in CH2Cl2 at 293 K

Extent of π-conjugation in ligands, dimers and polymers

Normalized emission spectra of P1–P4 in CH2Cl2 at 293 K
All the polymers show weak and broad red photoluminescence at room temperature (Fig. 4). In general, red luminescent polymers often suffer from a low emission efficiency since red emissive chromophores are normally prone to aggregation in the solid state and are highly susceptible to concentration quenching [57]. Due to the unstructured nature of their emission bands and the donor–acceptor nature in these systems, their emissions are most probably originated from the charge transfer excited states but not the ligand-centered π-π* excited state. In all cases, no triplet emission was observed which manifests the result of energy gap law for narrow-gap polyplatinaynes [58] and it is also in agreement with the observed emission lifetimes in the nanosecond regime [40]. Hence, we ascribe the ligand-dominating singlet excited state instead of the triplet state to contribute to the photoinduced charge separation in the energy conversion for the polymers.

J-V curves of solar cells with P1–P4:PCBM (1:4, w/w) active layers under simulated AM1.5 solar irradiation
3.3 Photovoltaic Properties
Since the light-induced intramolecular electron transfer could easily occur from donor to acceptor through the π-conjugation which favors photocurrent generation and the photoelectronic energy conversion in photovoltaic devices, PSCs with BHJ configuration were fabricated by using each of P1–P4 as electron donor and PCBM as electron acceptor (Fig. 5 and Table 3). The hole collection electrode consisted of indium tin oxide (ITO) with a spin-coated poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) layer, while Al served as the electron collecting electrode. Figure 5 shows the J-V curves of PSCs with P1–P4:PCBM (1:4, w/w) active layers under simulated AM1.5 solar irradiation. Absorption spectra of the neat films of P1–P4 blended with PCBM (1:4, w/w) are shown in Fig. S7, and as compared to the absorption spectra of P1–P4 in solution, they exhibit a wider coverage of the visible spectral range. The higher energy peak in the blend film is less pronounced because of the dramatically enhanced absorption in the UV region by the addition of PCBM [59].
The PSCs based on P3 and P4 show over 60% higher in Jsc than that of P1 and P2, while the PCE is notably increased when aldehyde group is replaced by the dicyanovinyl group. A higher PCE of 1.35% can be obtained for P4 with Voc = 0.79 V, Jsc = 4.47 mA cm−2 and FF = 0.38 under illumination of an AM 1.5 solar cell simulator in a 1:4 (P4:PCBM) blend ratio. It indicates that a higher PCE would be obtained if a stronger acceptor is attached to the side chain. This is also consistent with the weaker PL intensity in P3 and P4 with respect to that in P1 and P2 [60]. Addition of one more thiophene ring on both sides along the main chain can also increase PCE progressively from P1 to P2 and P3 to P4.
4 Conclusion
In conclusion, a set of new platinum-containing polymetallayne polymers functionalized with electron-rich triphenylamine backbone and different electron acceptor in the side chain have been successfully synthesized via Sonogashira-type dehydrohalogenation or Knoevenagel condensation reaction. All of them exhibit good solubility in common organic solvents and good thermal stability and show a broad absorption in the visible region. The energy level and bandgap of the polymers could be effectively tuned by changing the strength of the acceptor groups in the side chain. The open-circuit voltage (Voc), short-circuit current density (Jsc) and the fill factor (FF) values of the device with P4 as electron donor are 0.79 V, 4.47 mA cm−2 and 0.38, respectively, resulting in a PCE of 1.35%. A marked increase in the Jsc and PCE can be observed in P3 and P4 (stronger strength acceptor) relative to P1 and P2 (weaker strength acceptor) at the same blend ratio. We believe that if a stronger cyano acceptor was attached to the side chain, a lower bandgap polymer with an ideal energy level would be obtained, and a higher PCE could be anticipated, which provides valuable information for the production of new light-harvesting polymer materials for electrical power generation. Attempts will be continued to further improve the PCE in the future.
Data availability
No datasets were generated or analysed during the current study.
References
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger, Science 270, 1789–1791 (1995)
C.J. Brabec, N.S. Sariciftci, J.C. Hummelen, Adv. Funct. Mater. 11, 15–26 (2001)
K.M. Coakley, M.D. Mcgehee, Chem. Mater. 16, 4533–4542 (2004)
W.-Y. Wong, Macromol. Chem. Phys. 209, 14–24 (2008)
B.C. Thompson, J.M.J. Frechet, Angew. Chem. Int. Ed. 47, 58–77 (2008)
S. Günes, H. Neugebauer, N.S. Sariciftci, Chem. Rev. 107, 1324–1338 (2007)
J. Chen, Y. Cao, Acc. Chem. Res. 42, 1709–1718 (2009)
M. Helgesen, R. Sondergaard, F.C. Krebs, J. Mater. Chem. 20, 36–60 (2010)
C. Cui, Y. Li, Energy Environ. Sci. 12, 3225–3246 (2019)
N.S. Sariciftci, L. Smilowitz, A.J. Heeger, F.A. Wudl, Science 258, 1474–1478 (1992)
J.J.M. Halls, C.A. Walsh, N.C. Greenham, E.A. Marseglia, R.H. Friend, S.C. Moratti, A.B. Holmes, Nature 376, 498–500 (1995)
J.J.M. Halls, K. Pichler, R.H. Friend, S.C. Moratti, A.B. Holmes, Appl. Phys. Lett. 68, 3120–3122 (1996)
M.M. Wienk, J.M. Kroon, W.J.H. Verhees, J. Knol, J.C. Hummelen, P.A. van Hal, R.A.J. Janssen, Angew. Chem. Int. Ed. 42, 3371–3375 (2003)
M. Reyes-Reyes, K. Kim, D.J. Carroll, Appl. Phys. Lett. 87, 083506 (2005)
W. Ma, C. Yang, X. Gong, K. Lee, A. Heeger, Adv. Funct. Mater. 15, 1617–1622 (2005)
M.C. Scharber, D. Wuhlbacher, M. Koppe, P. Denk, C. Waldauf, A.J. Heeger, C.L. Brabec, Adv. Mater. 18, 789–794 (2006)
J. Peet, J.Y. Kim, N.E. Coates, W.L. Ma, D. Moses, A.J. Heeger, G.C. Bazan, Nat. Mater. 6, 497–500 (2007)
Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li, L. Yu, J. Am. Chem. Soc. 131, 56–57 (2008)
J. Hou, H.-Y. Chen, S. Zhang, G. Li, Y. Yang, J. Am. Chem. Soc. 130, 16144–16145 (2008)
H.Y. Chen, J.H. Hou, S.Q. Zhang, Y.Y. Liang, G.W. Yang, Y. Yang, L.P. Yu, Y. Wu, G. Li, Nat. Photonics 3, 649–653 (2009)
J. Hou, Z. Tan, Y. He, C. Yang, Y. Li, Macromolecules 39, 4657–4662 (2006)
Y. Hou, Y. Chen, Q. Liu, M. Yang, X. Wan, S. Yin, A. Yu, Macromolecules 41(41), 3114–3119 (2008)
C. Duan, W. Cai, F. Huang, J. Zhang, M. Wang, T. Yang, C. Zhong, X. Gong, Y. Cao, Macromolecules 43, 5262–5268 (2010)
J. Roncali, P. Leriche, A. Cravino, Adv. Mater. 19, 2045–2060 (2007)
Y. Wang, E. Zhou, Y. Liu, H. Xi, S. Ye, W. Wu, Y. Guo, C.-A. Di, Y. Sun, G. Yu, Y. Li, Chem. Mater. 19, 3361–3363 (2007)
E.-J. Zhou, Z. Tan, C.-H. Yang, Y.-F. Li, Macromol. Rapid Commun. 27, 793–798 (2006)
F. Huang, K.-S. Chen, H.-L. Yip, S.K. Hau, O. Acton, Y. Zhang, J. Luo, A.K.-Y. Jen, J. Am. Chem. Soc. 131, 13886–13887 (2009)
Y. Shirota, T. Kobata, N. Noma, Chem. Lett. 18, 1145–1148 (1989)
A. Higuchi, K. Ohnishi, S. Nomura, H. Inada, Y. Shirota, J. Mater. Chem. 2, 1109–1110 (1992)
Y. Shirota, J. Mater. Chem. 10, 1–25 (2000)
Y. Shirota, J. Mater. Chem. 15, 75–93 (2005)
W. Zhang, Z. Fang, M. Su, M. Saeys, B. Liu, Macromol. Rapid Commun. 30, 1533–1537 (2009)
Y. Zou, G. Sang, W. Wu, Y. Liu, Y. Li, Synth. Met. 159, 182–187 (2009)
X. Wang, M. Nakao, K. Ogino, H. Sato, H. Tan, Macromol. Chem. Phys. 202, 117–125 (2001)
M. Zhang, X. Ma, H. Zhang, L. Zhu, L. Xu, F. Zhang, C.-S. Tsang, L.Y.S. Lee, H.Y. Woo, Z. He, W.-Y. Wong, Chem. Eng. J. 430, 132832 (2022)
Y. Jin, Y. Zhang, Y. Liu, J. Xue, W. Li, J. Qiao, F. Zhang, Adv. Mater. 31, 1900690 (2019)
M. Qian, R. Zhang, J. Hao, W. Zhang, Q. Zhang, J. Wang, Y. Tao, S. Chen, J. Fang, W. Huang, Adv. Mater. 27, 3546–3552 (2015)
T. Wang, R. Sun, M. Shi, F. Pan, Z. Hu, F. Huang, Y. Li, J. Min, Adv. Energy Mater. 10, 2000590 (2020)
L. Xu, C.-L. Ho, L. Liu, W.-Y. Wong, Coord. Chem. Rev. 373, 233–257 (2018)
W.-Y. Wong, X.-Z. Wang, Z. He, A.B. Djurišić, C.-T. Yip, K.-Y. Cheung, H. Wang, C.S.K. Mak, W.-K. Chan, Nat. Mater. 6, 521–527 (2007)
W.-Y. Wong, C.-L. Ho, Acc. Chem. Res. 43, 1246–1256 (2010)
Y. Wei, B. Wang, W. Wang, J. Tian, Tetrahedron Lett. 36, 665–668 (1995)
Y. Wei, Y. Yang, J.-M. Yeh, Chem. Mater. 8, 2659–2666 (1996)
A. Yassar, C. Videlot, A. Jaafari, Sol. Energy Mater. Sol. Cells 90, 916–922 (2006)
J. Chatt, B. L. Shaw, J. Chem. Soc. 4020–4033 (1959).
G.B. Kauffman, L.A. Teter, Inorg. Synth. 7, 245–249 (1963)
S. Takahashi, Y. Kuroyama, K. Sonogashira, N. Hagihara, A convenient synthesis of ethynylarenes and diethynylarenes. Chem Inf. 11(47), 627–630 (1980)
W.-Y. Wong, C.K. Wong, G.L. Lu, K.W. Cheah, J.X. Shi, Z. Lin, J. Chem. Soc. Dalton Trans. 24, 4587–4594 (2002)
W.-Y. Wong, C.K. Wong, G.L. Lu, A.W.M. Lee, K.W. Cheah, J.X. Shi, Macromolecules 36, 983–990 (2003)
J. Lewis, N.J. Long, P.R. Raithby, G.P. Shields, W.-Y. Wong, M. Younus, J. Chem. Soc. Dalton Trans. 22, 4283–4288 (1997)
C.-L. Ho, C.-H. Chui, W.-Y. Wong, S.M. Aly, D. Fortin, P.D. Harvey, B. Yao, Z. Xie, L. Wang, Macromol. Chem. Phys. 210, 1786–1798 (2009)
C.-L. Ho, W.-Y. Wong, J. Organomet. Chem. 691, 395–402 (2006)
Y.L. Zhong, A. Midya, Z. Ng, Z.-K. Chen, M. Daenen, M. Nesladek, K.P. Loh, J. Am. Chem. Soc. 130, 17218–17219 (2008)
Q. Wang, W.-Y. Wong, Polym. Chem. 2, 432–440 (2011)
Q. Wang, Z. He, A. Wild, H. Wu, Y. Cao, U.S. Schubert, C.-H. Chui, W.-Y. Wong, Chem. Asian J. 6, 1766–1777 (2011)
W.-Y. Wong, K.-H. Choi, G.-L. Lu, J.-X. Shi, Macromol. Rapid Commun. 22, 461–465 (2001)
S.D. Cummings, R. Eisenberg, J. Am. Chem. Soc. 118, 1949–1960 (1996)
J.S. Wilson, N. Chawdhury, M.R.A. Al-Mandhary, M. Younus, M.S. Khan, P.R. Raithby, A. Köhler, R.H. Friend, J. Am. Chem. Soc. 123, 9412–9417 (2001)
W.-Y. Wong, W.-C. Chow, K.-Y. Cheung, M.-K. Fung, A.B. Djurišić, W.-K. Chan, J. Organomet. Chem. 694, 2717–2726 (2009)
W. Xu, M. Zhang, X. Ma, X. Zhu, S.Y. Jeong, H.Y. Woo, J. Zhang, W. Du, J. Wang, X. Liu, F. Zhang, Adv. Funct. Mater. 33, 2215204 (2023)
Acknowledgements
W.-Y. W. thanks the financial support from the RGC Senior Research Fellowship Scheme (SRFS2021-5S01), the National Natural Science Foundation of China (52073242), the Hong Kong Research Grants Council (PolyU 15307321), Research Institute for Smart Energy (CDAQ) and Miss Clarea Au for the Endowed Professorship in Energy (847S). Q. W. and J. L. thank the financial support from Chengdu University.
Funding
Open access funding provided by The Hong Kong Polytechnic University. RGC Senior Research Fellowship Scheme (SRFS2021-5S01), the National Natural Science Foundation of China (52073242), the Hong Kong Research Grants Council (PolyU 15307321), Research Institute for Smart Energy (CDAQ) and Miss Clarea Au for the Endowed Professorship in Energy (847S).
Author information
Authors and Affiliations
Contributions
Qiwei Wang: conceptualization, investigation, resources, supervision, writing-original draft; Lu Jiang: data curation, investigation, writing-original draft; Junlong Li: data curation, funding acquisition; Zelin Sun: data analysis, writing-review and editing; Wai-Yeung Wong: funding acquisition, project administration, resources, supervision, writing-review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Q., Jiang, L., Li, J. et al. Synthesis, Characterization and Photovoltaic Properties of Polyplatinaynes with Side Chain Functionalization by Different Electron-Accepting Group. J Inorg Organomet Polym (2024). https://doi.org/10.1007/s10904-024-03064-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10904-024-03064-w