
Abstract
Sex attractant pheromones are highly sensitive and selective tools for detecting and monitoring populations of insects, yet there has been only one reported case of pheromones being used to monitor protected species. Here, we report the identification and synthesis of the sex pheromone of a protected European moth species, Graellsia isabellae (Lepidoptera: Saturniidae), as the single component, (4E,6E,11Z)-hexadecatrienal. In preliminary field trials, lures loaded with this compound attracted male moths from populations of this species at a number of widely separated field sites in France, Switzerland, and Spain, clearly demonstrating the utility of pheromones in sampling potentially endangered insect species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sex pheromones or related attractants have been identified for several thousand insect species (Witzgall et al., 2004, El-Sayed, 2009), and use of pheromone-baited traps is now commonplace for detection, monitoring, and in some cases control of pest species. Because pheromone-baited traps usually are species-specific and sensitive, they can be particularly useful for detection of rare individuals or low-density populations. Thus, semiochemically-baited traps have been used widely for detection of incursions of quarantined insect pests, for monitoring the spread of exotic pests once they have been introduced, and for assessing the efficacy of eradication efforts (e.g., Mediterranean, oriental, Mexican, and melon fruit flies in the United States; painted apple moth in New Zealand; Kean and Suckling, 2005). Pheromone traps also have been used to detect low density populations of intentionally introduced biological control agents, to determine whether establishment has occurred (e.g., Cossé et al., 2005). However, to our knowledge, there has been only a single reported case of pheromones being used as tools for monitoring the distribution and abundance of protected species, for the saproxylic beetle Osmoderma eremita (Coleoptera: Scarabaeidae). This pheromone also serves as a kairomone for another rare species, the click beetle Elater ferrugineus (Coleoptera: Elateridae), which is a predator of O. eremita (reviewed in Larsson and Svensson, 2009).Footnote 1
The protected moth Graellsia isabellae (Graells 1849) (Lepidoptera: Saturniidae) is one of the most emblematic European insects. Because of its large size, attractive wing patterns, and restricted geographical distribution, it has become a symbol for several entomological and conservation organizations. It is univoltine, and adults fly at dusk from March to July in pine forests in the eastern part of Spain and the French Alps (Ylla Ullastre, 1997). Graellsia isabellae is protected by the Habitats Directive (the European Community initiative for an ecological network of special protected areas, known as “Natura 2000”), Bern Convention, Red Book of Lepidoptera, and Spanish and French red lists (Ministerio de Medio Ambiente, 2006; Ministère de l‘Écologie et du Développement Durable, 2008). Despite this protective legislation, the latest Red list of the IUCN considered G. isabellae as “data deficient” in terms of its current range and population density (IUCN, 2007). The natural history and ecology of G. isabellae has been studied (Ylla Ullastre, 1997, Chefaoui and Lobo, 2007, 2008, Vila et al., 2009), but conservation genetic studies have just started (Vila et al., 2010), and the current distribution and conservation status of the species needs to be updated because new populations have been discovered well beyond the previously known limits of its range (Ibáñez Gázquez et al., 2008).
Previous attempts at identifying the sex pheromone of G. isabellae were not successful, due in part to the very small amount of pheromone produced by the insects, and exacerbated by the relatively small number of individuals that were available for analysis of the pheromone gland contents (Zagatti and Malosse, 1998). A mass rearing program produced 245 females, but the amount of pheromone in a composite extract of the dissected glands from those females still proved to be below the threshold for detection by coupled gas chromatography-mass spectrometry (GC-MS). One fragment of information obtained from these efforts was that the antennae of male moths were extremely responsive to C16 aldehydes (Zagatti and Malosse, 1998), indicating that the pheromone was probably an aldehyde similar to those reported from some other saturniid species (Witzgall et al., 2004, El-Sayed, 2009).
As part of a larger effort to more effectively assess population sizes and delineate the range of G. isabellae, we describe here the identification and synthesis of the female-produced sex attractant pheromone of this insect. The results of preliminary field trials with the pheromone have shown that it is highly attractive to male moths from a number of widely separated populations, suggesting that pheromone-based methods will be effective for detection and sampling of this protected species, and will allow detailed assessments of its conservation status.
Methods and Materials
Insects
Pupae of G. isabellae galliaegloria Oberthür were obtained from a culture maintained by one of the authors (C.L.V.) at INRA Orléans. This culture has been out-crossed every year since the 1990s by mating laboratory-reared females with wild males to maintain the vigor of the laboratory colony. Larvae of G. isabellae galliaegloria were reared at INRA-Orléans in 2008, using a standard protocol (Collectif OPIE, 1998). Larval stock was collected from the upper Durance (Hautes Alpes, France). Pupae were over-wintered outdoors in a garden shed at INRA Orléans. Sixty female and 10 male pupae were sent to the quarantine facility at University of California, Riverside in April-May 2009 (USDA-APHIS permit # P526P-08-02961). Upon receipt in Riverside, pupae were separated by sex and placed in roughly 15 cm tall × 14 cm diam screen cages made from 6.3 mm mesh hardware cloth with 150 mm Petri plate bottoms. Each Petri plate bottom was covered with a dry paper towel with the pupae placed on the towel and the cages placed inside 4 l Ziploc® plastic bags with the tops open, and a saturated paper towel on top to ensure adequate humidity. These cages were then held in a Percival controlled environment chamber (#I-30BLL, Percival Scientific Inc., Perry, IA, USA) with 15°C night and 20°C daytime temperatures, and ≥60 % RH. The chamber was lit with two Phillips F20T12/CW lights on a 14:10 h L:D cycle with the lights turning off at 12:00 h. Newly emerged females were sensitive to disturbance and would frequently begin to deposit sterile eggs if disturbed, or if they were not mated during the first night. For these reasons, females were transferred gently from the cages holding groups of pupae to individual cages, and all pheromone collections were made from virgin, calling females during their first scotophase. Newly emerged males were either used immediately in coupled gas chromatography-electroantennogram detection (GC-EAD) assays, or were held at ca. 4°C in a Ziploc® plastic bag with a piece of damp paper toweling for up to 2 d. Male genitalia were clipped off prior to removing males from quarantine, as a required condition for their release from quarantine.
Solid Phase Micro-Extraction (SPME) Sampling of Sex Pheromone Glands
Crude extracts of pheromone were prepared from calling females by wipe sampling of extruded glands with 100 μm PDMS SPME fibers (Supelco, Bellefonte, PA, USA). Females were held in a controlled environment chamber under the same conditions as those for emergence (see above). Approximately 1–2 h after the start of scotophase, calling females were removed from the chamber into the darkened room. The SPME device was taped to the bench top so that both hands could be used to manipulate the insect, and approximately 5–7 mm of the SPME fiber was extruded. A microscope light source controlled with a rheostat was used to illuminate the fiber, and a small flashlight with red film covering the lens was used to confirm that females were indeed calling immediately prior to pheromone collection. Thus, a female was grasped firmly by the abdomen, with gentle pressure towards the tip of the abdomen to evert the sex pheromone gland (the scale-free segment just anterior to the ovipositor). Once exposed, this area of the abdomen was wiped gently over the SPME fiber, with all surfaces being wiped at least twice. Females then were placed individually into glassine envelopes and held in a refrigerator because they would usually begin to oviposit after this procedure. The loaded SPME fiber was analyzed by GC-EAD or GC-MS.
Solvent Extraction of Sex Pheromone Glands
Two composite solvent extracts of abdomen tips of calling females were made by removing glands and rinsing them in pentane. For the first composite extract, all 6 females were extracted on the same day, whereas the second extract from 7 females was made over a 4 d period, again using females during their first scotophase. Sex pheromone glands were removed approximately 1.5–2 h into the scotophase by gently forcing eversion of the sex pheromone gland, clamping the abdomen just anterior to the gland with forceps to maintain the pressure in the gland, then slicing the gland off with a razor blade such that the gland remained inflated in the forceps. The gland then was rinsed with gentle agitation in ca. 1 ml of clean pentane for 30 sec. These extracts were analyzed by coupled GC-EAD without concentration, and by GC-MS after concentration under a gentle stream of nitrogen.
GC-EAD and GC-MS Analyses
GC-EAD analyses were performed using DB-5 and DB-Wax columns (both 30 m × 0.25 mm i.d., 0.25 μm film; J&W Scientific, Folsom CA, USA). Helium was used for both the carrier and makeup gas. Temperature programs were 100°C for 1 min, 10°C per min to 275°C for DB-5 and 250°C for DB-Wax, hold for 60 min. SPME injections were made by desorbing the fiber in splitless mode for 60 sec prior to starting the run (injector temperature 250ºC). Solvent extracts (1 μl aliquots) were analyzed in splitless mode. The effluent from the columns was split using an ‘X’ cross, with half of the sample going to the FID detector and the other half to the EAD. The portion directed to the EAD was diluted in a humidified air stream prior to exposure to the antenna. Males used in GC-EAD analyses were removed from quarantine after the genitalia had been removed, allowed to warm to room temperature, and then a single antenna was removed by pulling it off at the scape. The terminal rami were removed with a razor blade, and the tip was excised to ensure good contact with the saline solution (7.5 g NaCl, 0.21 g CaCl2, 0.35 g KCl, and 0.20 g NaHCO3 in 1 l Milli-Q purified water) in the glass electrodes, which were fitted with an internal gold wire for connection to the amplifier. A single antennal preparation was used for as many as four runs. Kovat’s indices were calculated for unknowns and standards relative to blends of straight-chain hydrocarbons.
Syntheses of Authentic Standards
The syntheses of mixtures of (4E/Z, 6E,11E)- and (4E/Z,6E,11Z)-hexadecatrienals are described in the supporting online information.
Tetrahydrofuran was purified by distillation from sodium-benzophenone ketyl under argon. Unless otherwise specified, solutions of crude products were dried over anhydrous Na2SO4 and concentrated by rotary evaporation under reduced pressure. Crude products were purified by flash or vacuum flash chromatography on 230–400 mesh silica gel. 1H- and 13C-NMR spectra were recorded on a Varian INOVA-400 spectrometer (Palo Alto, CA, USA) (400 and 100.5 MHz, respectively), as CDCl3 solutions unless otherwise stated. Solvent extracts of reaction mixtures were dried over anhydrous Na2SO4 and concentrated by rotary evaporation under reduced pressure. Integer resolution mass spectra were obtained with a Hewlett-Packard (HP) 6890 GC (Avondale PA) interfaced to an HP 5973 mass selective detector, in EI mode (70 eV) with helium carrier gas. The GC was equipped with a DB5-MS column (25 m × 0.20 mm ID × 0.25 μ film).
4-Nonyn-1-ol (3)
A solution of THP-protected 4-pentyn-1-ol 1 (5.04 g, 30 mmol) and 25 mg triphenylmethane as an indicator in 50 ml THF was cooled to 0ºC, and treated dropwise with n-BuLi (2.56 M in hexanes, ~12.5 ml) until the solution began to turn orange-red, indicative of excess n-BuLi. The solution was stirred 20 min, the ice bath was removed, and butyl iodide (6.44 g, 35 mmol) was added by syringe over 5 min. The solution was refluxed 44 h under Ar, then quenched by pouring into cold 1 M NH4Cl. The mixture was extracted with hexane, and the hexane layer was washed with saturated brine, then dried and concentrated. The residue was purified by Kugelrohr distillation (oven temp ~105ºC, 0.05 mm Hg), yielding the protected alkynol 2 as a colorless oil (5.6 g, 83%). The product was dissolved in 20 ml MeOH, 0.25 g of para-toluenesulphonic acid (PTSA) was added, and the solution was stirred overnight at room temperature. The solution then was poured into 100 ml of 0.5 M aqueous NaOH, and the mixture was extracted 3 times with hexane. The combined hexane extracts were washed with brine, dried, and concentrated. The residue was purified by Kugelrohr distillation, taking off a preliminary fraction containing THP-protected MeOH (bp < 40ºC at 1 mm Hg), followed by distillation of the remaining 4-nonyn-1-ol 3 (3.13 g, 94%; oven temp < 100ºC at 1 mm Hg). 1H NMR: δ 3.76 (t, 2H, J = 6.2 Hz), 2.26–2.31 (tt, 2H, J = 7.2, 2.4 Hz), 2.12–2.17 (m, 2H), 1.74 (overlapped tt, 2H, J ~ 7, 7 Hz), 1.67 (br. s, OH), 1.35–1.5 (m, 4H), 0.91 (t, 3H, J = 7.2 Hz). 13C NMR: δ 13.8, 15.7, 18.6, 22.2, 31.3, 31.8, 62.3, 79.4, 81.3 ppm. The 13C spectrum matched that previously reported (Poleschner and Heidenreich, 1995).
(4Z)-Nonen-1-ol (4)
A 1 M solution of NaBH4 was prepared by dissolving 0.4 g NaBH4 in a mixture of 9.5 ml EtOH and 0.5 ml 2 M aqueous NaOH, and the slightly cloudy solution was filtered. In a separate flask, a solution of NiOAc2•4 H2O (1.0 g, 4 mmol) was prepared in 50 ml of degassed 95% EtOH under Ar, giving a clear green solution. A balloon of hydrogen gas was attached to the flask, the Ar line was removed, and with vigorous stirring, 1 ml of the NaBH4 solution (1 mmol) was added dropwise, producing a black suspension with the evolution of H2. The mixture was stirred 10 min, then 0.8 ml of ethylenediamine was added. After stirring an additional 10 min, 4-nonyn-1-ol 3 (2.90 g, 21.4 mmol) was added by syringe. The mixture was stirred until all the starting material had been consumed (3 h), then flushed with Ar, and filtered with suction through a pad of Celite®, rinsing well with EtOH. The violet-colored solution was concentrated to ~10 ml, then poured into 100 ml 1 M HCl and extracted 3 times with hexane. The combined hexane layers were washed with saturated aqueous NaHCO3 and brine, then dried and concentrated. The residue was purified by Kugelrohr distillation (oven temp to 75ºC, 0.5 mm Hg), yielding (4Z)-nonen-1-ol 4 (2.76 g, 91%) as a colorless oil. 1H NMR: δ 5.39 (m, 2H), 3.66 (t, 2H, J = 6.4 Hz), 2.13 (m, 2H), 2.06 (m, 2H), 1.64 (overlapped dt, 2H, J ~ 6.5 Hz), 1.49 (br. s, OH), 1.28–1.36 (m, 4H), 0.90 (t, 3H, J = 7.2 Hz). 13C NMR: δ 14.2, 22.5, 23.8, 27.1, 32.1, 32.9, 62.9, 129.0, 121.0 ppm. The 1H NMR data agreed with that previously reported (Joshi et al., 1984).
(4Z)-1-Bromononene (5)
Methanesulphonyl chloride (1.89 g, 16.5 mmol) was added dropwise to a solution of (4Z)-nonen-1-ol 4 (2.13 g, 15 mmol) and Et3N (2.02 g, 20 mmol) in 50 ml CH2Cl2 at 0ºC. The resulting slurry was stirred at 0ºC for 2.5 h, then poured into ice-water. The organic layer was separated and washed with 1 M HCl, saturated aqueous NaHCO3, and brine, then dried and concentrated. The residue was taken up in 40 ml acetone, LiBr (3.9 g, 45 mmol) was added, and the mixture was stirred at 50ºC overnight, by which time all the mesylate had been consumed. The slurry was cooled and poured into 250 ml water, and the product was extracted with hexane. The hexane layer was washed with brine, dried, and concentrated, and the residue was purified by Kugelrohr distillation (oven temp ≤75ºC at 5.5 mm Hg), yielding the bromide 5 (2.76 g, 74%) as a colorless oil. 1H NMR: δ 5.45 (dtt, 1H, J = 10.2, 7.2, 1.4 Hz), 5.31 (dtt, 1H, J = 10.8, 7.6, 1.6 Hz), 3.42 (t, 2H, J = 6.6 Hz), 2.20 (m, 2H), 2.06 (m, 2H), 1.92 (overlapped tt, 2H, J ~ 7 Hz), 1.31–1.37 (m, 4H), 0.91 (t, 3H, J = 6.8 Hz). 13C NMR: δ 14.2, 22.5, 25.9, 27.2, 32.1, 32.9, 33.6, 127.6, 132.0 ppm. The 1H and 13C NMR data were in agreement with those previously reported (Ducoux et al., 1992).
(6Z)-Undecen-1-yne (6)
A dry flask under Ar was loaded with lithium acetylide-ethylenediamine complex (0.92 g, 10 mmol), and dry DMSO (10 ml) and NaI (150 mg, 1 mmol) were added, followed by dropwise addition of (4Z)-1-bromononene 5 (1.44 g, 7 mmol). The mixture was stirred 1 h at room temperature, then quenched by pouring into 100 ml water. The mixture was extracted twice with hexane, and the combined hexane layers were washed with brine, dried, and concentrated. The residue was purified by Kugelrohr distillation (oven temp ~80ºC, 12 mm Hg), giving 0.87 g (83%) of (6Z)-undecen-1-yne 6 as a colorless oil. 1H NMR: δ 5.42 (dtt, 1H, J = 10.8, 7.2, 1.2 Hz), 5.33 (dtt, 1H, J = 10.8, 7.2, 1.2 Hz), 2.20 (td, 2H, J = 7.0, 2.4 Hz), 2.16 (overlapped td, 2H, J ~ 7.2 Hz), 2.05 (m, 2H), 1.96 (t, 1H, J = 2.4 Hz), 1.59 (overlapped tt, 2H, J ~ 7.4 Hz), 1.34 (m, 2H), 0.91 (t, 3H, J = 7.0 Hz). 13C NMR: δ 14.2, 18.1, 22.5, 26.4, 27.1, 28.7, 32.1, 68.4, 84.7, 128.5, 131.3 ppm. The 1H and 13C NMR spectra agreed with those previously reported (Asao et al., 2005).
(4E,6E,11Z)-Hexadecatrien-1-ol (11)
A dry flask was charged with bis(cyclopentadienyl)-zirconium hydridochloride (1.2 g, 4.65 mmol; Alfa Aesar, Ward Hill MA, USA) and flushed thoroughly with Ar. The flask was cooled in an ice-bath, THF was added, and the slurry was stirred 15 min, followed by dropwise addition of (6Z)-undecen-1-yne 6 (0.60 g, 4 mmol). The mixture was shielded from light, and stirred at 0ºC for 20 min, then warmed to room temperature and stirred for 3.5 h. The resulting solution was then cooled to 0ºC again, and a solution of iodine (1.07 g, 4.2 mmol) in 5 ml THF was added dropwise until the brown color of excess iodine persisted. The solution was stirred at 0ºC for 45 min, then warmed to room temperature and quenched by pouring into 50 ml of 2 M sodium thiosulphate solution. Hexane (100 ml) was added, and the slurry was mixed thoroughly, then filtered with suction through a pad of Celite®. The organic layer was separated and washed sequentially with sodium thiosulphate solution and brine, then dried and concentrated. The residue was purified by vacuum flash chromatography on silica gel, eluting with pentane. The purified (1Z,6Z)-1-iodoundecadiene 7, contaminated with (6Z)-1,6-undecadiene (from reduction instead of iodination of the zirconium intermediate), was concentrated and used immediately in the next step. 1H NMR: δ 6.52 (dt, 1H, J = 14.4, 7.2 Hz), 5.99 (dt, 1H, J = 14.4, 1.4 Hz), 5.36 (m, 2H), 1.99–2.1 (m, 6H), 1.46 (overlapped tt, 2H, J ~ 7.4 Hz), 1.30–1.37 (m, 4H), 0.91 (t, 3H, J = 7.2 Hz).
A dry flask was charged with bis(cyclopentadienyl)zirconium hydridochloride (1.7 g, 6.6 mmol) and evacuated and refilled with Ar 5 times. The flask was cooled to ~ − 15ºC in an ice-salt bath, and 7.5 ml of dry CH2Cl2 were added. The mixture was stirred 15 min, then t-butyldimethylsilyl-protected 4-pentyn-1-ol 8 (1.19 g, 6 mmol) was added dropwise. The mixture was stirred 30 min, warmed to 0ºC and stirred 30 min, then warmed to room temperature and stirred 30 min. The solvent then was removed under vacuum, and the alkenylzirconium intermediate 9 was taken up in 15 ml THF. A solution of anhydrous ZnCl2 (0.94 g, 7 mmol) in 10 ml THF was added, and the solution was cooled to −78ºC and degassed under vacuum, then warmed to 0ºC, followed by addition of a solution of (1Z,6Z)-1-iodoundecadiene 7 (0.83 g, ~3 mmol) and tetrakis(triphenylphosphine)palladium in 10 ml THF. The mixture was stirred for 1 h, warmed to room temperature, and stirred until the iodide had been completed consumed (~2 h). The reaction was quenched by pouring into 50 ml saturated NH4Cl, and the resulting mixture was extracted twice with hexane. The combined hexane extracts were washed with brine, and treated with anhydrous Na2SO4 and decolorizing charcoal for 1 h with stirring. The mixture was filtered with suction through a pad of Celite®, then concentrated. The residue was purified by vacuum flash chromatography on silica gel, eluting with 5% EtOAc in hexane. The purified product 10 was concentrated, and the residue was stirred in a mixture of 6 ml THF, 6 ml AcOH, and 3 ml H2O at room temperature overnight to hydrolyze the silyl protecting group. The mixture then was poured into 100 ml water and extracted with 75 ml hexane. The hexane layer was washed sequentially with saturated aqueous NaHCO3 and brine, dried, and concentrated. The residue was purified by vacuum flash chromatography, eluting with 15% EtOAc in hexane, yielding (4E,6E,11Z)-hexadecatrien-1-ol 11 (0.57 g, 80%, 96% isomerically pure by GC). Attempted recrystallization from hexane at −20ºC was not successful, and so the product was Kugelrohr distilled (0.55 g, 78%; bp ~ 110ºC, 0.1 mm Hg) to remove traces of silica gel. 1H NMR: δ 6.04 (m, 2H), 5.58 (m, 2H), 5.36 (m, 2H), 3.67 (t, 2H, J = 6.8 Hz), 2.16 (m, 2H), 2.0–2.1 (m, 6H), 1.67 (overlapped tt, 2H, J ~ 7 Hz), 1.4–1.48 (m, 2H), 1.29–1.36 (m, 4H), 0.90 (t, 3H, J = 7 Hz). 13C NMR: δ 14.2, 22.6, 26.9, 27.1, 29.1, 29.6, 32.2, 32.3, 32.5, 62.7, 129.6, 130.5 (2C), 131.2, 131.4, 132.8 ppm. The spectra agreed with those previously reported (Tomida and Fuse, 1993).
(4E,6E,11Z)-Hexadecatrienal (12)
Oxalyl chloride (0.13 ml, 1.5 mmol) was added by syringe to 3 ml of dry CH2Cl2 cooled under Ar to −78ºC in a dry-ice acetone bath. A solution of dry dimethylsulfoxide in CH2Cl2 (0.225 ml, 3.36 mmol in 1 ml) was added dropwise over 15 min. The mixture was stirred 20 min, then (4E,6E,11Z)-hexadecatrien-1-ol 11 (0.236 g, 1 mmol) in 1 ml of CH2Cl2 was added over 5 min, and the mixture was slowly warmed to ~ −45ºC over 30 min, and stirred at that temperature for 30 min. The mixture was cooled to −78ºC again, 1 ml of Et3N was added dropwise, and the resulting mixture was slowly warmed to room temperature over ~1 h. The mixture then was poured into ice-water and extracted with hexane. The hexane layer was washed successively with saturated NaHCO3 and brine, then applied directly to a column of silica gel (20 ml), eluting the column with 10% EtOAc in hexane. Fractions containing the aldehyde were combined and purified further by Kugelrohr distillation, yielding the aldehyde as a pale yellow liquid (171 mg, 73%). The aldehyde was diluted immediately with hexane to a 5% solution, ~5 mg BHT were added as stabilizer, and the solution was sealed in brown glass ampoules flushed with nitrogen to minimize degradation. NMR spectra were run in deuteroacetone to minimize problems with acid-catalyzed trimerization of the long-chain aldehyde that might result from traces of DCl in the typical NMR solvent, CDCl3. 1H NMR (deuteroacetone): δ 9.71 (t, 3H, J = 1.6 Hz), 5.95–6.08 (m, 2H), 5.53–5.62 (m, 2H), 5.29–5.39 (m, 2H), 2.51 (br. t, 2H, J ~ 6.8 Hz), 2.34 (br. quart., 2H, J ~ 6.9 Hz), 2.0–2.1 (m, 6H, partially obscured by deuteroacetone peak), 1.38–1.45 (m, 2H), 1.24–1.34 (m, 4H), 0.87 (t, 3H, J = 7.2 Hz). 13C NMR: δ 13.56, 22.28, 25.06, 26.59, 26.85, 31.99, 32.04, 43.10, 129.47, 130.17 (2C), 130.73, 131.52, 132.69, 201.63 ppm. One carbon signal appeared to be obscured by the deuteroacetone signals from 28.6–29.7 ppm. The NMR spectra were in general agreement with those previously reported in CDCl3 solvent (Tomida and Fuse, 1993). MS (EI, 70 EV): 234 (M+, 2), 216 (1), 190 (7), 177 (6), 163 (7), 159 (9), 150 (23), 134 (38), 121 (38), 119 (47), 107 (26), 95 (51), 93 (53), 91 (48), 81 (59), 79 (100), 67 (86), 55 (55), 41 (57).
Field Trials
Lures were prepared at INRA Orléans from 11-mm red rubber septum lures (Wheaton Scientific, Millville, NJ, USA) loaded with heptane solutions (100 μl, 1 mg/ml) of the synthesized pheromone that had been shipped by courier to INRA from UC Riverside. Loaded septa were placed in a 20-ml glass vial and kept in a freezer or an ice-chest when not in use. Five different experiments were set up to test the efficiency of the lures and compare the attraction of male moths to pheromone lures, light traps, or calling females:
Experiment 1
Lures were tested on 19 May 2009 in the Spanish Pyrenees (San Juan de la Pena, Huesca province, 42.51675°N, 0.69056°W, 1100 m above sea level [a.s.l.]). Two pheromone-treated or untreated control septa were placed in one mesh cage [16 cm H × 32 cm W × 16 cm D]. In total, 4 control and 4 treatment cages (a total of 16 septa) were suspended from branches of Pinus sylvestris, one of the moth’s main host plants, ~0.5 m above ground. Cages were spaced approximately 2 m apart, and were monitored from 21:30 till 23:00 (males mostly fly at dusk). The temperature ranged from 25.4°C at the beginning of the experiment to 11.4°C at the end. Responding males were collected with a net, tissues were sampled non-lethally for DNA analyses, as part of a conservation genetics study, and adults were then released (Vila et al., 2010).
At the same time, we set up a light trap consisting of a night collecting sheet (Bioquip, CA, USA) with a 175 W mercury vapor light mounted on a tripod on one side of the sheet. The light trap was placed 280 m away from the lures (42.51824°N, 0.69341°W, 952 m a.s.l.).
Experiment 2
On 20 May 2009, lures were tested in the Spanish Central Sierra of Guadarrama (Dehesas de Cercedilla 40.76768°N, 4.07006°W, 1375 m a.s.l.). The experimental design was the same as described above, but temperature ranged from 21.9°C to 15.5°C. At the same time, we set up a light trap and 2 calling females (2 d old) in one cage one km away from the lures (40.75798°N, 4.07148°W, 1300 m a.s.l.).
Experiment 3
On 27 and 28 May 2009, lures were tested in the Swiss Alps (Schallberg, Canton du Valais, 46° 29.594′N, 8° 02.581′E, 1364 m a.s.l.). A single pheromone treated septum was nailed to the trunk of a P. sylvestris tree 1.5 m above the ground. We placed a total of 7 unmated females a few days old in 5 cages at approximately 15 m from the lure. The experiment started at 21:30 and finished at 23:00. On May 27, temperature was 11ºC when the experiment started and 4ºC at the end. On May 28, temperature was 11ºC at the beginning, and 6ºC when the experiment was terminated.
Experiment 4
On 3 June 2009, we set up lures at the Spanish Natural Park of Puebla de San Miguel (Valencia province, 40.04325°N, 1.06284°W, 1717 m a.s.l.). A single pheromone treated septum was nailed to the trunk of a P. sylvestris tree, 1.5 m from the ground. We set a light trap (one 175 W lamp ) approximately 140 m from the lure. We monitored both lures and light trap from 21:30 till 23:00. Males were collected with a net, tissues were non-lethally sampled for DNA analyses, and adults were then released.
Experiment 5
On 11 June 2009, we nailed two pheromone treated septa to two pine trees 800 m apart, in the same area as experiment 4 (40.07914°N 1.10293°W and 40.07209°N 1.10473°W). In addition, we set a sticky trap baited with an untreated septum (control), nailed to a pine trunk on the same pine tree used for experiment 4. We simultaneously placed the lures and the control 1.5 m above the ground at 21:30. Different observers monitored the septa until 23:00.
Results
Identification of the Pheromone
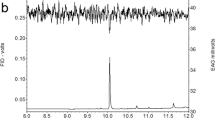
Analyses of SPME wipe samples and solvent extracts of dissected pheromone glands by GC-EAD showed that only a single trace component in the extracts elicited responses from antennae of male moths (Fig. 1). This compound had a Kovats index (KI) of 1861 on a relatively nonpolar DB-5 column, and 2383 on a polar DB-WAX column. In GC-MS analyses, the active component showed a small molecular ion at m/z 234 (1% of base peak), and a base peak at m/z 79, for a possible molecular formula of C16H26O, consistent with a C16 trienal. Comparison of its KI values with those of model compounds suggested that the compound might contain a conjugated diene and an isolated carbon-carbon double bond. Specifically, the KI of the model compound (6E,11Z)-hexadecadienal was 80 KI units less than that of the unknown on the DB-5 column, suggesting that at least one of the double bonds was conjugated either with another carbon-carbon double bond or with the aldehyde. Furthermore, the KI values of the model compound (5Z,7Z,11Z)-hexadecatrienal (1855 on DB-5 and 2383 on DB-WAX) were similar to those of the unknown, providing support for a C16 trienal with a conjugated diene structure. An attempt to determine the position of the diene by derivatization with MTAD failed due to the limited amount of pheromone gland extract available, and the trace amount of pheromone in the extracts. Furthermore, the much stronger responses of antennae of male moths to model compounds with aldehyde vs. alcohol functional groups provided corroborating evidence for the presence of a C16 trienal rather than the corresponding tetraenol that would be required by the molecular weight of m/z 234.

Coupled gas chromatography-electroantennogram analysis of a SPME wipe sample of everted pheromone gland of a female Graellsia isabellae. Top trace is the GC trace; bottom, inverted trace is the response from the male moth antenna. DB-5 column, 100ºC/1 min, 10ºC/min to 275ºC for 60 min
From the limited number of pheromones known from moths in the same subfamily (Saturniinae), two species (Antheraea polyphemus and A. pernyi) had been shown to produce (6E,11Z)-hexadecadienal as a pheromone component (Kochansky et al., 1975, Bestmann et al., 1987, respectively), and a third species, the Ailanthus silkmoth Samia cynthia ricini, produced (4E,6E,11Z)-hexadecatrienal (Bestmann et al., 1989). Thus, with no other hard information as to the positions and geometries of the double bonds in the unknown, we synthesized four of the eight possible isomers of this compound. The retention indices of three of these four isomers on the DB-5 column did not match that of the unknown, excluding them from further consideration (KI values: 4E,6E,11E: 1840; 4Z,6E,11E, 1871; 4Z,6E,11Z, 1837). However, the retention time of synthetic (4E,6E,11Z)-hexadecatrienal exactly matched that of the unknown on both the DB-5 and the DB-WAX columns, and the mass spectra of the synthetic and natural compounds were also a good match, providing a tentative identification of the pheromone as (4E,6E,11Z)-hexadecatrienal. Further confirmation of this identification was obtained from field bioassays (see below).
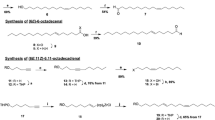
Synthesis of (4E,6E,11Z)-hexadecatrienal
Four of the eight possible isomers of 4,6,11-hexadecatrienal were synthesized as isomeric pairs, using Wittig reactions to assemble the conjugated diene structures (see Supplementary Online Information). Having determined that the (4E,6E,11Z)-isomer matched the insect-produced compound in all respects, we then developed a route to that specific isomer (Fig. 2). A key starting material, 4-nonyn-1-ol, was not commercially available, and so it was assembled by alkylation of THP-protected 4-pentyn-1-ol with butyl iodide (83%) (Buck and Chong, 2001). After deprotection (94%), the resulting alkynol 3 was stereoselectively reduced to (Z)-4-nonen-1-ol with P-2 nickel and hydrogen, and then converted to (Z)-4-nonenyl bromide 5 by treatment with methanesulphonyl chloride and pyridine followed by LiBr in acetone (67% over 2 steps). Reaction of bromide 5 with lithium acetylide-ethylene diamine complex in DMSO (Sonnet and Heath, 1980) then produced enyne 6 (83%). Treatment with bis(dicyclopentadienyl)zirconium hydridochloride followed by iodine (Zeng et al. 2004) stereospecifically gave the key (E)-vinyl iodide 7. A second application of hydridozirconation to t-butyldimethylsilyl-protected 4-pentyn-1-ol 8 gave the alkenylzirconium intermediate 9. This was converted in situ to the corresponding zinc organometallic, and palladium-catalyzed coupling of this intermediate with iodide 9 completed the assembly of the E,E-conjugated diene system (Ribe et al., 2000). Removal of the alcohol’s t-butyldimethylsilyl protecting group in aqueous acid gave trienol 11 in 78% yield from enyne 6. Swern oxidation of alcohol 11 completed the synthesis, and after purification, the resulting aldehyde was immediately diluted in hexane and sealed in brown ampoules to minimize degradation.

Stereoselective synthesis of (4E,6E,11Z)-hexadecatrienal
Field Bioassays
Experiment 1
A total of 20 males were attracted to the treatment cages, whereas no males were attracted to cages containing untreated control septa. Cages with pheromone-treated septa attracted males from 21:48 till 22:30. No males were attracted with the light trap.
Experiment 2
A total of 11 males were attracted by the treated lures. No males were attracted by the controls, by the light trap, or by caged calling females.
Experiment 3
During the course of this experiment conducted over two successive evenings, no males flew towards the caged females; all males observed flew rapidly to the pheromone lures. On May 27, we collected 10 males (between 21:36 and 22:30), all of which were marked by writing a number in the upper-side right forewing, then released at 23:00. On May 28, we collected 34 males, seven of which were recaptures from the previous evening. The first male (non-recaptured) arrived only one min after setting out the pheromone lure.
Experiment 4
We collected 48 males attracted by the pheromone lure, whereas only one male was attracted by the light trap. Males showed no hesitation in approaching the lure, indicating that the single component was both necessary and sufficient to obtain attraction.
Experiment 5
Four males were attracted by the lure set at site 40.07209°N 1.10473°W, and seventeen males were attracted to the lure deployed at site 40.07914°N 1.10293°W. Interestingly, individuals flew off after a few minutes. No males came to the control lure.
Discussion
As suggested by the strong EAG responses elicited by aldehyde standards in preliminary work by Zagatti and Malosse (1998), the pheromone of G. isabellae proved to be a triunsaturated aldehyde, (4E,6E,11Z)-hexadecatrienal. In GC-EAD analyses of SPME wipe samples or solvent extracts of pheromone glands of female moths, this compound elicited large responses from antennae of male moths. Antennae of males showed negligible responses to any other compounds in the extracts, suggesting that the pheromone consists of only a single compound. This was supported by the consistent attraction of males to lures containing the trienal as a single component. The synthetic pheromone lures proved to be more attractive than either light traps (experiments 1, 2, and 4) or calling females (experiments 2 and 3). The increased attraction to the synthetic pheromone lures in comparison to virgin females may have been a result of dose, or the normal calling behavior of females may have been disturbed by handling.
We often observed the first male/s flying readily to the lures a few minutes (1–5) after deploying the septa. In experiments where the septa were nailed to the trunk of a tree, males stayed on the septum itself most of the time, or walked and flew close by or on the tree trunk before flying off a few minutes later. The fact that males that were initially attracted to lures abandoned their search for a female and flew off after a few minutes suggests that other, short-range signals such as contact sex pheromones may be required in order to elicit the full sequence of mating behaviors and/or that males that cannot quickly find the pheromone source (i.e., a female) allocate further efforts to searching for a more accessible mate.
(4E,6E,11Z)-Hexadecatrienal had been previously reported as a pheromone component for another saturniid species, the Ailanthus silkmoth Samia cynthia ricini (Bestmann et al., 1989), and the related dienal, (6E,11Z)-hexadecadienal had been found in other members of the family (Antherea polyphemus, Kochansky et al., 1975; Antherea pernyi Bestmann et al., 1987). Although (4E,6E,11Z)-hexadecatrienal had been synthesized previously, all reported syntheses used reactions with low stereoselectivity, such as Wittig reactions (Bestmann et al., 1989, Tomida and Fuse, 1993) or Claisen orthoester rearrangements (Singh et al., 1992). Thus, we developed an improved synthesis based on zirconium and zinc organometallics, with a key step being the palladium-catalyzed coupling reaction between alkenylzirconium intermediate 9 and vinyl iodide 7. In our hands, this proved to be highly stereoselective, giving the desired product in 96% isomeric purity.
Although the pheromone was identified from females belonging to subspecies G. isabellae galliaegloria (French Alps), the synthesized pheromone attracted males from a total of three subspecies: G. isabella paradisea Marten (experiment 1: Central Spanish Pyrenees), G. i. isabellae Graells (experiment 2: Sierra Guadarrama; experiments 4&5: Valencia), and G. i. galliaegloria (experiment 3: Swiss Alps).
Previous to this work, field surveys for this species had been carried out using light traps and calling female moths. The identification and testing of the pheromone described here has demonstrated how the pheromone can be used to dramatically increase the sensitivity of survey methods while simultaneously decreasing the time and effort required to carry out surveys. Having the pheromone in hand will allow us to survey areas where the species is potentially present (Chefaoui and Lobo, 2007, 2008), demonstrate absence with far greater certainty than previously possible, and identify isolated populations of potentially high conservation value.
The success of this project also has shown that problems associated with working with the pheromones of rare or endangered lepidopteran species are manageable, for several reasons. First, the extensive literature on lepidopteran pheromones allows predictions as to likely pheromone structures based on the taxonomic placement of species. Second, with care, it is possible to collect pheromone samples by SPME wipe sampling with no damage to calling females, so that the females can be used subsequently in captive breeding programs. Alternatively, if it is necessary to make solvent extracts of pheromone glands, the sensitivity of current analytical instruments requires that only a few females be sacrificed. Third, field surveys can be carried out with observers or video cameras simply watching pheromone lures, with no insects being caught, so surveys should have minimal impact on moth populations. If responding males are indeed caught with nets, if done carefully, the males sustain minimal damage, as evidenced by our being able to use males captured in this way for mark-recapture studies. For all these reasons, the identification and use of pheromones would seem to be a practical and efficient method of detecting and monitoring rare and endangered species.
Notes
Whereas nothing has been published to date, over the past decade, J.G.M. and J.S.M. have supplied pheromone lures to the Nature Conservancy for monitoring populations of several endangered species in the United States.
References
Asao, N., Sato, K., Menggenbateer, and Yamamoto, Y. 2005. AuBr3- and Cu(OTf)2-catalyzed intramolecular [4 + 2] cycloaddition of tethered alkynyl and alkenyl enynones and enynals: a new synthetic method for functionalized polycyclic hydrocarbons. J. Org. Chem. 70:3682–3685.
Bestmann, H. J., Attygalle, A. B., Brosche, T., Erler, J., Platz, H., Schwarz, J., Vostrowsky, O., Wu, C.-H., Kaissling, K. E., and Chen, T.-M. 1987. Identification of three sex pheromone components of the female Saturniid moth Antheraea pernyi (Lepidoptera: Saturniidae). Z. Naturforsch. C. 42:631–636.
Bestmann, H. J., Attygalle, A. B., Schwarz, J., Garge, W., Vostrowsky, O., and Tomida, I. 1989. Pheromones 71. Identification and synthesis of female sex pheromone of Eri-silkworm, Samia cynthia ricini (Lepidoptera: Saturniidae). Tetrahedron Lett. 30:2911–2914.
Buck, M. and Chong, J. M. 2001. Alkylation of 1-alkynes in THF. Tetrahedron Lett. 42:5825–5827.
Chefaoui, R. M. and Lobo, J.M. 2007. Assessing the conservation status of an Iberian moth using pseudo-absences. J. Wildl. Manage. 71:2507–2516.
Chefaoui, R. M. and Lobo, J. M. 2008. Assessing the effects of pseudo-absences on predictive distribution model performance. Ecol. Modell. 210:478–486.
Collectif O.P.I.E. 1998. Contribution à la connaissance de Graellsia isabellae galliaegloria Oberthur (Lépidoptère, Attacidae), connu uniquement en France. Rapport d’Études de l’OPIE 3:1–36. Office pour les Insectes et leur Environnement, Guyancourt, France.
Cossé, A. A., Bartelt, R. J., Zilkowski, B. W., Bean, D. W., and Petroski, R. J. 2005. The aggregation pheromone of Diorhabda elongata, a biological control agent of saltcedar (Tamarix spp.): identification of two behaviorally active components. J. Chem. Ecol. 31:657–670.
Ducoux, J.-P., Le Ménez, P., Kunesch, N., Kunesch, G., and Wenkert, E. 1992. An efficent and stereoselective synthesis of insect pheromones by way of nickel-catalyzed Grignard reactions. Syntheses of gossyplure and pheromones of Eudia pavonia and Drosophila melanogaster. Tetrahedron 48:6403–6412.
El-Sayed, A. 2009. The Pherobase. http://www.pherobase.com.
Ibáñez Gázquez, S., Nevado Ariza, J. C., and Ylla Ullastre, J. 2008. Graellsia isabelae (Graells, 1849), una nueva especie para la fauna lepidopterológica de Almería (España) (Lepidoptera: Saturniidae): SHILAP. Rev. Lepidopterol. 36:427–430.
International Union for the Conservation of Nature. 2007. IUCN Red List of Threatened Species. URL http://www.iucnredlist.org.
Joshi, N. N., Mamdapur, V. R., and Chadha, M. S. 1984. Stereoselective and versatile approach for the synthesis of gossyplure and its components. Tetrahedron 40:3285–9.
Kean, J. M. and Suckling, D. M. 2005. Estimating the probability of eradication of painted apple moth from Auckland. N. Z. Plant Protection 58:7–11.
Kochansky, J., Tette, J., Taschenberg, E. F., Cardé, R. T., Kaissling, K.-E., and Roelofs, W. L. 1975. Sex pheromone of the moth, Antheraea polyphemus. J. Insect Physiol. 21:1977–1983.
Larsson, M. C. and Svensson, G. P. 2009. Pheromone monitoring of rare and threatened insects: exploiting a pheromone–kairomone system to estimate prey and predator abundance. Conserv. Biol. 23:1516–1525.
Ministère de l’Écologie et du Développement durable (2008) ESPECES ANIMALS. 2008. 1075—Isabelle de France (Graellsia isabellae) Cahiers d’habitats. URL http://www.ecologie.gouv.fr/IMG/natura2000/habitats/pdf/tome7/1075.pdf.
Ministerio de Medio Ambiente. 2006. Graellsia isabellae. Catálogo Nacional de Especies Amenazadas. URL http:// www.mma.es/secciones/biodiversidad/especies_amenazadas/catalogo_especies/artropodos/pdf/INV38.pdf.
Poleschner, H. and Heidenreich, M. 1995. 13C NMR chemical shifts of unbranched alk-2-yn-1-ols, 3-alkyn-1-ols and ‘internal’ alkyn-1-ols. Mag. Res. in Chem. 33:917–21.
Ribe, S., Kondru, R. K., Beratan, D. N., AND Wipf, P. 2000. Optical rotation computation, total synthesis, and stereochemistry assignment of the marine natural product pitiamide A. J. Am. Chem. Soc. 122:4608–4617.
Singh, V., Vig, R., Mohini, V., Trehan, I. R., and Kad, G. L. 1992. Syntheses of (4E,6E,11Z)-hexadecatrienyl acetate and (4E,6E,11Z)-hexadecatrienal, female sex pheromone of Eri-silkworm, Samia cynthia ricini (Lepidoptera: Saturniidae). Proc. Indian Acad. Sci. (Chem. Sci.) 104:57–60.
Sonnet, P. E. and Heath, R. R. 1980. Stereospecific synthesis of (Z,Z)-11,13-hexadecadienal, a female sex pheromone of the navel orangeworm, Amyelois transitella (Lepidoptera: Pyralidae). J. Chem. Ecol. 6:221–228.
Tomida, I. and Fuse, T. 1993. Preparation of four geometrical isomers of the Eri-silk moth pheromone, (11Z)-4,6,11-hexadecatrienals, and their effect toward male Eri-silk moths. Biosci. Biotech. Biochem. 57:648–652.
Vila, M., Auger-Rozenberg, M. A., Goussard, F., and Lopez-Vaamonde, C. 2009. Effect of non-lethal sampling of DNA on life history traits of the protected moth Graellsia isabelae (Lepidoptera: Saturniidae). Ecol. Entomol. 34: 356–362.
Vila, M. , Marí-Mena, N., Yen, S.-H., and Lopez-Vaamonde, C. 2010 Characterization of ten polymorphic microsatellite markers for the protected Spanish moon moth Graellsia isabelae (Lepidoptera: Saturniidae). Conserv. Genet. 11: 1151–1154.
Witzgall, P., Lindblom, T., Bengtsson, M., and Tóth, M. 2004. The Pherolist. www-pherolist.slu.se.
Ylla Ullastre, J . 1997. Història Natural del Lepidòpter Graellsia isabelae (Graells, 1849). Institut d’Estudis Catalans, Arxius de les Seccions de Sciencies CXVI, Barcelona , Spain. 232 p.
Zagatti, P. and Malosse, C. 1998. Étude de la phéromone sexuelle et mise au point d’une phéromone de synthèse. (ed. OPIE). In : Contribution à la connaissance de Graellsia isabelae galliaegloria Oberthur (Lepidoptera, Attacidae) connu uniquement en France. Rapport d'études de l’OPIE 3:26–27.
Zeng, X., Zeng, F., Negishi, E.-I. 2004. Efficient and selective synthesis of 6,7-dehydrostipiamide via Zr-catalyzed asymmetric carboalumination and Pd-catalyzed crosscoupling of organozincs.Org. Lett. 6:3245–3248.
Acknowledgments
We thank Francis Goussard, David Lees, Enrique Murria, Joaquin Baixeras, Guillermo Fernández, Piedad Lincango, and José Sevilla for assistance in the field. We thank Raimondas Mozuraitis for his work and comments on previous attempts to identify the pheromone using SPME. This work was performed under permits from all environmental authorities concerned, and financially supported by the Spanish Ministry of Education and Science (research grant CGL2007-63549/BOS and Acción Integrada HF2007-0055). MV was funded by Xunta de Galicia (Isidro Parga Pondal programme). NMM was funded by a FPI grant (BES-2008-002571).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 136 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Millar, J.G., McElfresh, J.S., Romero, C. et al. Identification of the Sex Pheromone of a Protected Species, the Spanish Moon Moth Graellsia isabellae . J Chem Ecol 36, 923–932 (2010). https://doi.org/10.1007/s10886-010-9831-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10886-010-9831-1