
Abstract
Autoimmune lymphoproliferative syndrome (ALPS) is a rare genetic disorder featuring chronic lymphadenopathy, splenomegaly, cytopenias, and increased lymphoma risk. Differentiating ALPS from immunodeficiencies with overlapping symptoms is challenging. This study evaluated the performance and the diagnostic yield of a 15-gene NGS panel for ALPS at Cincinnati Children’s Hospital Medical Center. Samples from 802 patients submitted for ALPS NGS panel were studied between May 2014 and January 2023. A total of 62 patients (7.7%) had a definite diagnosis: 52/62 cases (84%) showed 37 unique pathogenic/likely pathogenic germline FAS variants supporting ALPS diagnosis (6.5%, 52/802). The ALPS diagnostic yield increased to 30% in patients who additionally fulfilled abnormal ALPS immunology findings criteria. 17/37 (46%) diagnostic FAS variants were novel variants reported for the first time in ALPS. 10/802 cases (1.2%) showed diagnostic findings in five genes (ADA2, CTLA4, KRAS, MAGT1, NRAS) which are related to autoimmune lymphoproliferative immunodeficiency (ALPID). Family studies enabled the reclassification of variants of unknown significance (VUS) and also the identification of at-risk family members of FAS-positive patients, which helped in the follow-up diagnosis and treatment. Alongside family studies, complete clinical phenotypes and abnormal ALPS immunology and Fas-mediated apoptosis results helped clarify uncertain genetic findings. This study describes the largest cohort of genetic testing for suspected ALPS in North America and highlights the effectiveness of the ALPS NGS panel in distinguishing ALPS from non-ALPS immunodeficiencies. More comprehensive assessment from exome or genome sequencing could be considered for undefined ALPS-U patients or non-ALPS immunodeficiencies after weighing cost, completeness, and timeliness of different genetic testing options.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a rare primary immunodeficiency disorder of defective Fas-mediated apoptosis (restimulation-induced cell death), featuring chronic lymphadenopathy, splenomegaly, cytopenias, and increased lymphoma risk. While typically seen in childhood, symptoms can manifest at any age. The diagnostic approach to suspicious patients tends to be broad and often includes imaging studies as well as invasive diagnostic procedures, such as the biopsy of lymph nodes and/or other tissues. The typical biomarker for ALPS is an increased number of a normally uncommon population of “double-negative TCRαβ + CD3 + CD4- CD8- T cells” (DNTs); other characteristic laboratory abnormalities include defective lymphocyte apoptosis, elevated levels of interleukin (IL) IL-10, IL-18, vitamin B12, and soluble FAS-ligand (sFASL) [1].
The molecular basis for ALPS was gradually deciphered with the advances in sequencing and molecular technology. In 1995, heterozygous pathogenic variants in FAS were first reported in children with ALPS [2, 3]. A year later, Wu et al. reported a heterozygous 84-bp deletion in exon 4 of FASLG in a patient originally thought to have systemic lupus erythematosus (SLE) [4], but later was considered more consistent with ALPS type Ib [5]. Later publications showed monoallelic loss-of-function variants in FASLG are tolerated. ALPS-FASLG (ALPS type Ib) is likely caused by biallelic loss-of-function variants [6]. In 1999, Wang et al. identified heterozygous pathogenic variants in CASP10 in ALPS type II [5], but its pathogenicity has been debated [7]. In 2004, somatic pathogenic FAS variants were reported in ALPS [8]. In 2009, a genetic diagnosis was recommended to be part of the ALPS diagnostic criteria and the ALPS classification was recommended to be based on genes with pathogenic variants detected [9]. Recently, germline heterozygous FADD mutations, coupled with a somatic loss of heterozygosity (LOH) in DNTs, have been reported to cause ALPS [10] and was classified as ALPS-FADD [7]. Whereas the biallelic mutations in FADD are associated with immunodeficiency 90 with encephalopathy, functional hyposplenia, and hepatic dysfunction (Online Mendelian Inheritance in Man (OMIM) #613759), which is not an ALPS variant. It has been recently proposed to restrict the ALPS classification to conditions with clear evidence of FAS signaling defect and resulting ALPS specific clinical and laboratory manifestations [7]. Autoimmune lymphoproliferative immunodeficiency (ALPID) was proposed as a clinical “umbrella” term that includes conditions that may initially present with an ALPS-like phenotype and biomarkers at the borders, but show additional non-ALPS symptoms later on [7].
With more ALPS patients having genetic defects identified and reported, we developed a clinical FAS gene sequence test in 2005, then FASLG and CASP10 sequencing tests in 2009. With the advance of the next-generation sequencing (NGS) technology, in 2014, we developed an NGS panel that includes FAS, FASLG, FADD (monoallelic mutations), and CASP10, as well as genes associated with top differential diagnoses of ALPID, including CASP8, CTLA4, FADD (biallelic mutations), ITK, MAGT1, PRKCD, STAT3, KRAS, LRBA, NRAS, RASGRP1, and ADA2 (CECR1).
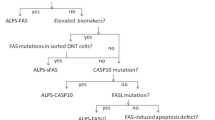
In this paper, we present a retrospective study of 802 patients undergoing the ALPS NGS panel at Cincinnati Children’s Hospital Medical Center (CCHMC). We report on the primary molecular and immune characteristics of ALPS and some ALPID and the difficulties encountered during genetic testing. Furthermore, we assess and summarize the clinical utility of our ALPS testing algorithm (Suppl. Figure 1), which aligns with the testing guideline outlined in the 2009 NIH international workshop for the genetic diagnosis of ALPS patients [9].
Methods
Patient’s Enrollment
A retrospective study of 802 individuals who had an ALPS NGS panel performed at CCHMC from May 2014 to January 2023 was conducted to evaluate the performance of the test. The patient’s clinical phenotypes based on review of the requisitions and/or chart review, the pathogenicity of variants, and, where available, the laboratory findings of the ALPS Immune-panel and Fas-mediated lymphocyte apoptosis were evaluated and summarized (Table 1). Testing was ordered by clinicians from various institutions.
DNA Extraction and NGS test
Genomic DNA was extracted from peripheral blood. NGS was conducted on 9 genes (CASP8, CASP10, FADD, FAS, FASLG, ITK, KRAS, MAGT1, NRAS) for 548 cases from May 2014 to October 2019 or 15 genes (additional genes: ADA2, CTLA4, LRBA, PRKCD, RASGRP1, and STAT3) for 254 cases from November 2019 to January 2023. Variants were classified according to the ACMG guidelines [11]. Somatic variants with > = 5% allele fraction in FAS were targeted for cases performed after November 2019. Supplementary material.
Flow Cytometry ALPS Immune-Panel
An ALPS Immune-panel was completed for 101 of 254 patients tested for ALPS NGS panel from November 2019 to January 2023. Due to the change of the institutional LIMS system, we had limited access to the ALPS Immune-panel results completed before November 2019. Therefore, we only reviewed the ALPS Immune-panel results for patients with abnormal ALPS NGS panel (30 patients).
This flow cytometry panel interrogates 4 parameters suggestive of ALPS (TCRαβ DNTCs > 2% or > 68 cells/ul; B220 + TCRαβ DNTCs > 60%; CD3 + CD25+ / HLA DR ratio < 1.0; CD27 + B cells < 15%), based on published results [12,13,14]. A score system was utilized with 4/4 as consistent with ALPS; 3/4 as suspicious for ALPS; 2/4 as not typical for ALPS; and 1/4 or 0/4 as not consistent for ALPS. Peripheral lymphocyte subsets were evaluated from whole blood using an 8-color immunostaining panel (lyse and wash procedure), a FACS Canto II flow cytometer (BD) equipped with three lasers (blue, red, violet), FACSDiva software (BD), and a large panel of RUO mAbs and fluorochromes variously combined (all BD). This panel is clinically available (https://www.testmenu.com/cincinnatichildrens/Tests/660966).
Fas-mediated Apoptosis Test
An in vitro Fas-mediated apoptosis test was conducted on 23 out of 254 patients tested for ALPS NGS panel from November 2019 to January 2023. Due to the Institutional LIMS change, we only reviewed the apoptosis results for patients with abnormal ALPS NGS panel (11 patients) for cases completed before November 2019.
In short, peripheral blood mononuclear cells were suspended in complete RPMI 1640 medium and T cells were activated with Concanavalin A. Activation was assessed using CD3, CD8 and CD25. After 4 days, activated T cells were washed and then cultured in medium supplemented with recombinant human IL-2 for 7 days. Expanded T cells were then plated in duplicate in 96-well plates and treated with agonistic anti-Fas antibody (APO-1-3, Alexis Biochemicals) and Protein A in the presence of IL-2 to evaluate Fas-mediated lymphocyte apoptosis. Twenty-one hours after treatment, cells were stained with propidium iodide and analyzed by flow cytometry. Cell death was quantified as % cell loss = (1-(% viable cells, treated / % viable cells, untreated)) x 100. Percent Cell Loss was reported from a 500 ng/mL concentration of APO-1-3 and Protein A. Fas function was defined as defective when cell survival was out of the reference range (68–93%). This test is clinically available (https://www.testmenu.com/cincinnatichildrens/Tests/660968).
Results
Demographic Findings in 802 Patients with Suspected ALPS
Between May 2014 and January 2023, 802 patients with suspected ALPS from North America were referred from different institutions and underwent ALPS NGS testing. The characteristics of the cohort are summarized in Tables 1; 63% (504/802) were male and 37% (294/802) were female. The median age at testing was 12 years (ranging from 1 month to 76 years). Of the 548 patients tested from May 2014 to October 2019 with race/ethnicity information, almost 40% (219/548) were European American.
The Diagnostic Yield of ALPS NGS Panel in 802 Patients
The diagnostic yield of ALPS NGS panel in 802 patients with suspected ALPS was 7.7% (62/802). Of the 62 positive cases, 52/62 (84%) had FAS pathogenic/likely pathogenic variants supporting the diagnosis of ALPS. Of the 52 FAS positive cases, 73% (38/52) were male and 27% (14/52) were female, with a male predominance as previously suggested [15].
The remaining 10/62 (16%) had pathogenic/likely pathogenic variants in CLTA4, MAGT1, KRAS, ADA2, and NRAS (Fig. 1A).

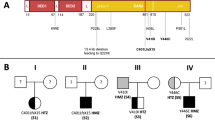
(A) Pie charts reporting the frequency of diagnostic variants identified in the 802-patient cohort, grouped by gene, and the frequency of FAS variants grouped by domain. (B) Schematic diagram of the locations of 37 diagnosis germline variants in 52 patients with respect to the predicted domains of FAS. Each dot represents one patient and novel variants are bolded. The intracellular death domain (AA230-314 region) is highlighted in orange-red color. CID, Calcium Inducing Domain; DD, Death Doman
No diagnostic variants were detected in FASLG and CASP10, suggesting ALPS-FASLG and ALPS-CASP10 are rare.
66 cases had variants of unknown significance (VUS).
Among 62 positive cases and 66 cases with VUS (total 128 cases), 23 cases (18%) underwent follow-up family studies in our institution (Table 1). All 23 families were tested for the variants in FAS. Some representative family study results were shown in Fig. 3.
Genetic Findings in FAS
52 out of 802 (6.5%) patients had a heterozygous pathogenic or likely pathogenic variant in FAS. 37 unique FAS variants were detected in 52 cases, with 11 (30%) variants shared by multiple patients (Fig. 1B; Table 2). 20/37 (54%) variants have been previously reported in patients with ALPS. 17/37 (46%) variants were novel variants which had not been previously reported in any ALPS patients (bold in Fig. 1B; Table 2). Of these 52 cases, 22 (42%) were heterozygous for a truncating variant or a missense variant in the extracellular domain (ECD) or a variant resulting in the transmembrane (TM) domain-coding exon 6 skipping. The effect of these variants was reported to be either decreased FAS expression or localization, or inability to bind FAS ligand with reduced FAS ligand-induced apoptosis [16]. 30/52 (58%) cases were heterozygous for variants in the intracellular domain (ICD), of which, 17 (57%) were with missense variants in the death domain, which plays a key role in the death-inducing signaling [16] (Fig. 1B; Table 2). Most of the 17 novel variants were truncating variants or missense variants in the death domain, except c.197G>C p.(Gly66Ala). This variant is in ECD and has not been reported in the literature, but a different variant at the same nucleotide position resulting in a different amino acid change, c.197G>A p.(Gly66Asp), has been reported in a patient with ALPS [17], suggesting the functional importance of this locus. p.(Gly66Ala) is predicted to be deleterious (REVEL score of 0.854) and was classified as likely pathogenic based on the ACMG variant classification criteria [11].
The ALPS NGS panel also tested for somatic FAS variants with allele fraction (AF) > = 5% for 254 cases processed from November 2019 to January 2023. However, no somatic FAS variants were detected.
Genetic Findings in Other Genes Related to ALPID
Given the similar clinical features associated with ALPS and other lymphoproliferative disorders, our ALPS NGS panel also includes genes associated with the top differential diagnoses of ALPID. There were 10 cases that showed a diagnostic result in five genes associated with ALPID (Fig. 1A; Table 3), including one homozygous variant in ADA2 that causes vasculitis, autoinflammation, immunodeficiency, and hematologic defects [18]; three heterozygous variants in CTLA4 that cause immune dysregulation with autoimmunity, immunodeficiency, and lymphoproliferation [19, 20]; three likely heterozygous variants in KRAS or NRAS that cause RAS-associated autoimmune leukoproliferative disorder (RALD) [19, 21, 22]; and three hemizygous variants in MAGT1 that causes immunodeficiency [23, 24]. Additionally, there were three cases with a heterozygous variant but an uncertain diagnostic result because the genes are associated with autosome recessive diseases and a second variant was not detected (Suppl. Table 1).
Correlation of the ALPS Immune-panel Findings and Fas-mediated Apoptosis with Disease-causing Variants Detected in ALPS NGS Panel
Out of the 52 FAS-mutant cases, 32 cases had ALPS Immune-panel completed, either by our institution or elsewhere (per the provider report). We summarized the affected domains of FAS variants detected in these 32 cases and their ALPS Immune-panel results in Fig. 2A and Suppl. Table 2. Although not knowing whether the patients received immunosuppressive drugs at the time of testing, all but two of the 17 cases with pathogenic/likely pathogenic ICD variants had abnormal ALPS Immune-panel results, reporting “consistent with ALPS” or “suspicious for ALPS”. However, only 9/13 (69%) cases with ECD variants had an abnormal ALPS Immune-panel. This result is consistent with the reported incomplete penetrance of the ECD germline FAS pathogenic variants, especially those with a truncating effect [13, 16]. The four cases with ECD FAS variants and “not typical” or “not consistent” ALPS Immune-panel results all had frameshift variants, predicted to result in haploinsufficiency (Pts 2, 4, 6, 16 in Table 2).

ALPS phenotypes and FAS genotypes correlation. (A) The correlation of ALPS immune-panel results with variants in different FAS protein domains. The numbers around the chord diagram represent the number of patients. (B) Cloud map shows the high frequency of patients’ phenotypes. Splenomegaly counts in 31 of a total 52 FAS-positive patients, elevated double negative T (DNT) cells counts in 28 of 52 (Including cases with the abnormal ALPS immune-panel results), and lymphadenopathy counts in 24 of 52 (Including enlarged lymph nodes and lymphadenopathy). (C) Heatmap showed the percentage of different phenotypes in FAS-positive patients, grouped by FAS variant’s location in terms of the extracellular and intracellular domains
Where available, we also evaluated the patients’ phenotypes and the phenotype-genotype correlation from the 52 FAS-positive cases. Splenomegaly, lymphadenopathy, and elevated DNTs were the top three phenotypes in these patients (Fig. 2B). Consistently, patients with ECD FAS pathogenic/likely pathogenic variants showed more reduced penetrance for the characteristic clinical phenotype of ALPS, compared to the ICD variants (Fig. 2C).
Of the 52 FAS-positive cases, ten cases underwent Fas-mediated apoptosis testing. One case failed due to technical issues, and 9/9 (100%) cases with a valid apoptosis test showed decreased Fas-mediated apoptosis (Table 2). This result is consistent with germline FAS pathogenic variants invariably having defective Fas-mediated apoptosis. This confirmed previous findings that the penetrance of abnormal in vitro Fas-mediated apoptosis in ALPS-FAS is approximately 100%, regardless of the variant location [13, 25].
Among the 10 cases positive for other ALPID, 8 cases had ALPS Immune-panel results (Table 3). While the majority of cases (6/8) showed “not consistent” or “not typical” results, two patients (Pts 60 and 61 in Table 3) showed “suspicious for ALPS” results, and both of them were hemizygous for pathogenic variants in MGAT1. These findings underscore the significance of genetic testing in facilitating the accurate diagnosis of ALPS.
Correlation of the ALPS Immune-panel and Fas-mediated Apoptosis in Patients with VUS in FAS
Of the 66 cases who had a VUS in the ALPS NGS Panel, 31/66 (47%) cases had ALPS Immune-panel tests performed in our institution or elsewhere (per the provider report). Five cases had FAS VUS and “consistent” or “suspicious” results for ALPS Immune-panel (Table 4). Three of these cases had Fas-mediated apoptosis tests performed and found two were abnormal. Specifically, Pt 64 is heterozygous for a previously unreported synonymous variant, c.390A>G p.(Lys130=). This variant is predicted to enhance a cryptic splice donor site 4-bp upstream of this change. Per the provider’s report, this patient met the clinical criteria for ALPS, although the available information for this variant was not sufficient to classify it as likely pathogenic.
The other patient (Pt 66) is heterozygous for a missense variant located in the intracellular death domain, c.710C>T p.(Ala237Val). This variant has not been published. It has been reported in the gnomAD database in one heterozygote in all populations. REVEL score is 0.545 suggesting uncertain prediction on variant effect. Family studies showed his mother and sibling did not carry this variant, but his father was not available for testing.
Performance of the ALPS NGS Panel in Patients with Suspected ALPS and Abnormal Immune-panel and/or Fas-mediated Apoptosis Results
Due to the lower (7.7%) diagnostic rate observed in our 802-patient cohort compared to the reported attribution of FAS, FASLG, and CASP10 to ALPS [26], we systematically reviewed cases with the ALPS Immune-panel and/or the Fas-mediated apoptosis tests performed at our institution from November 2019 to January 2023. This was correlated with ALPS NGS sequencing results, to evaluate the diagnostic rate of ALPS NGS panel on patients with suspected ALPS and abnormal ALPS immunology results.
During the specified period, a total of 254 ALPS NGS panel cases were processed at our institution. 101/254 (40%) cases also underwent testing with the ALPS Immune-panel. 81/101 (80%) cases showed “inconsistent” or “not typical” results for ALPS. 20/101 (20%) were “suspicious for ALPS” or “consistent with ALPS”. In these twenty cases, 6 (30%) were positive for FAS pathogenic or likely pathogenic variants that supported the ALPS diagnosis (Suppl. Table 3), which was much higher than the diagnostic rate from the ALPS “immunology-unscreened” cohort.
Two patients were heterozygous for previously unreported VUS in FAS (Pts 63, 64). Of the remaining twelve patients with abnormal ALPS Immune-panel but no detected FAS variants, six underwent Fas-mediated apoptosis, revealing abnormal findings in two cases (Pts 70, 76 in Suppl. Table 3). These findings suggest that patients with suspected ALPS might be classified with ALPS-U or had pathogenic variants in the deep intronic regions/UTR regions in FAS, FADD, FASLG, or CASP10, which were not covered/detected in this genetic test. They could also have a somatic FAS variant with less than 5% VAF, or somatic FADD, FASLG or CASP10 which was not targeted in this panel. Additional genetic testing might be warranted.
Discussion
ALPS predominantly is caused by FAS mutations, either germline or somatic in DNT cells [15, 27]. In this study, we examined 802 patients with suspected ALPS referred to our institution for ALPS NGS panel from May 2014 to January 2023. The diagnosis yield of this ALPS NGS panel was 7.7%, which was lower than the reported attribution of FAS, FASLG, and CASP10 to ALPS [26]. This suggests that the ALPS NGS panel may have been utilized more as a screening test for patients with suspected ALPS, or a lymphoproliferative and/or autoimmune disorder in general, which was recently proposed to be termed as ALPID [7].
Having more complete clinical information, particularly regarding the fulfillment of ALPS criteria could potentially increase the genetic diagnosis rate, as shown by the 20 cases, with abnormal ALPS immunology testing results, evaluated between November 2019 and January 2023. In this smaller cohort, the diagnostic yield of the ALPS NGS panel was much higher at 30% when including cases with abnormal ALPS immunology criteria.
The nine exons of the FAS gene encode a signal sequence in exons 1–2 that is cleaved upon trafficking of the Fas protein to the cell surface, three cysteine-rich ECDs in exons 3–5, a TM in exon 6, and ICDs spanning exons 7–9 comprising the exon 9-encoded death domain that interacts with the intracellular apoptosis-inducing signal transduction pathway [28]. We detected variants across all of these regions with the greatest number of pathogenic variants detected in the intracellular death domain (Fig. 1). The reduced penetrance of ALPS phenotypes of ECD variants has also been observed in our data (Fig. 1; Families 3 and 7 in Fig. 3B).

Family study results. (A) c.794A>G; p.(Asp265Gly), was detected as de novo in one family, and in the other family, this variant segregated with disease. (B) other representative family study results. The black arrows indicate the probands. Circle = female, square = male. The legend for the keys is at the top of the figure
Variants in intron 5 and exon 6 are interesting and have been reported to result in skipping of exon 6 and producing a soluble Fas molecule that lacks the transmembrane domain [16, 29,30,31,32,33,34]. This soluble isoform blocks apoptosis induced by Fas. However, it has been proposed that haploinsufficiency, rather than the effect of an excessive production of soluble Fas, is the underlining disease mechanism [29]. This group of pathogenic variants is associated with a low penetrance of disease phenotype [29] like other variants in ECDs [16].
In our cohort, we detected three variants associated with exon 6 skipping in 5 probands, c.506-16A>G (Pt 18), c.568G>A which located at the last base of exon 6 (Pt 22), and c.506-3C>T (Pts 19, 20, 21). The c.506-16A>G and c.568G>A variants have previously been reported in patients with ALPS, both in a homozygous state [33] and in a heterozygous state, with cDNA sequencing showing in-frame skipping of exon 6 (p.Gly169_Trp189del). This results in a protein lacking the transmembrane domain [16].
Focusing on several illustrative cases; the c.506-16A>G variant was detected in a 15-year-old male with a clinical history of cardiac transplant, post-transplant lymphoproliferative disease (PTLD) with > 25% DNT cells, hepatosplenomegaly, enlarged lymph nodes, lymphoma, and sensorineural deafness (Pt 18, Table 2; Fig. 3B). Family studies showed this variant was inherited from his mother who had severe juvenile rheumatoid arthritis. His 17-year-old sister carries the variant and had left conductive hearing loss, atrial septal defect, heart palpitations, reduced exercise tolerance, and lymphadenopathy. His 12-year-old sister also carries this variant but without any relevant symptoms suggestive of ALPS. Family studies enabled the identification of at-risk family members and additional follow-up in this family.
The c.506-3C>T variant has not been previously reported in patients with ALPS, though a functional study using a minigene system showed it to lead to exon 6 skipping [30]. We detected c.506-3C>T in the heterozygous state in three male probands (Pts 19, 20, 21) with splenomegaly, red cell anemia, and thrombocytopenia as the common features. Patient 19 also had enlarged lymph nodes and reported abnormal ALPS Immune-panel. Patient 21 had neutropenia, intermittent rashes, and a paternal family history of ALPS. Though previously unreported in ALPS patients, c.506-3C>T is one of the top three common diagnostic variants detected in our cohort (Fig. 1B; Table 2).
It has been known that the most common genetic causes of ALPS are monoallelic pathogenic FAS variants followed by somatic FAS variants restricted to DNT cells [26]. Like ALPS-FADD, the combination of a germline FAS mutation and a somatic event (either mutations or LOH) impairing the second FAS allele (“2 hits”) has also been reported and accounts for the onset of clinical phenotype in ALPS, especially for ECD haploinsufficiency variants that have lower penetrance [35]. Detection of somatic FAS variants historically has been done by Sanger sequencing in sorted DNT cells. However, this procedure is expensive and technically challenging. NGS testing using DNA from whole blood has recently been reported to be able to detect somatic FAS mutations in seven ALPS patients [36] with FAS variant allele fraction ranging from 1.9 to 11.5% (median 5.9%). However, in our 254 cases from November 2019 to January 2023, in which the somatic FAS detection was included in the pipeline, we did not detect any somatic FAS variants. It is possible our somatic variant detection sensitivity (5%) is not high enough to detect these variants. For patients meeting ALPS criteria, with “consistent” or “suspicious” ALPS Immune-panel results, an abnormal Fas-mediated apoptosis, and a negative family history, further testing for somatic variants using sorted DNTC or NGS panel with higher sensitivity (< 5%) for somatic variants should be considered.
One limitation of the custom designed ALPS NGS panel is its lack of flexibility to add new genes to the panel. As additional genes associated with ALPID are identified, for example, PIK3CD [37, 38] and PIK3R1 [39, 40], regularly updating the custom NGS panel would increase the diagnostic yield. If not seeking somatic FAS variants, physicians could also consider ordering WES-based or WGS-based testing for patients with suspected ALPS as the sequencing cost continues to decrease. For patients who underwent genetic testing before the NGS approach was adopted, it might be worthwhile to retest, while re-analysis of WES-based testing might also be considered.
However, the number of VUS detected and reported may pose challenges to its clinical utility. Family studies to see whether the variant is de novo in the proband or whether the variant segregates with the disease in the family will provide additional evidence to upgrade or downgrade VUS. For example, the c.794A>G p.(Asp265Gly) variant detected in two independent families (Pts 44 and 45) was originally reported as VUS in 2016. Subsequent family studies revealed that it was de novo in one family and segregated with disease in another family (Fig. 3A). This additional information allowed us to upgrade this VUS to likely pathogenic in our database before this variant was reported in the literature [41, 42].
Importantly, complete clinical phenotypes and biological correlates, such as those obtained through specific ALPS-related studies (ALPS Immune-panel, cytokines, Vitamin B12, and Fas-mediated apoptosis) would be helpful to provide supporting evidence for variant classification and should be considered in the variant classification work up. Utilizing additional markers, such as CD38, HLA-DR, and CD45RA [13, 43, 44], to discriminate between ALPS-specific DNTs and DNTs not related to ALPS would also be helpful in differentiating ALPS from other ALPID and increasing the genetic diagnostic yield of ALPS.
Our study, while reporting novel genetic findings from a large cohort of patients with suspected ALPS, has its limitations. It is a retrospective study conducted at a single center, potentially limiting its generalizability. Most cases were ordered from outside institutions, resulting in limited information regarding the patient’s clinical history (collected from the patient’s requisitions). Not all patients had ALPS immune results and even for cases with a result, we did not know whether they were on immunosuppressive drugs, which may influence the ALPS Immune-panel results and potentially somatic variant detection. These factors all affect the diagnostic rate.
In summary, we report the investigation of an NGS targeted gene panel on a large cohort of patients with suspected ALPS. Identification of FAS or other ALPID gene mutations with this NGS strategy is a cost- and time-effective option to expedite the diagnosis and treatment of these patients avoiding further complications due to a delayed diagnosis, although most cases remain genetically undiagnosed. Additional functional tests, including RNA sequencing, and family studies on VUS may further resolve uncertain results.
Data Availability
Data is provided within the manuscript or supplementary information files.
References
Palmisani E, et al. Autoimmune lymphoproliferative syndrome (ALPS) disease and ALPS phenotype: are they two distinct entities? Hemasphere. 2023;7(3):e845.
Fisher GH, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–46.
Rieux-Laucat F, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–9.
Wu J, et al. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;98(5):1107–13.
Wang J, et al. Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98(1):47–58.
Maccari ME, et al. Revisiting autoimmune lymphoproliferative syndrome caused by Fas ligand mutations. J Allergy Clin Immunol. 2023;151(5):1391–e14017.
Magerus A, et al. Autoimmune lymphoproliferative immunodeficiencies (ALPIDs): a proposed approach to redefining ALPS and other lymphoproliferative immune disorders. J Allergy Clin Immunol. 2024;153(1):67–76.
Holzelova E, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351(14):1409–18.
Oliveira JB, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–40.
Pelle O, et al. Combined germline and somatic human FADD mutations cause autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2024;153(1):203–15.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Bleesing JJ, et al. TcR-alpha/beta(+) CD4(-)CD8(-) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol. 2001;100(3):314–24.
Bleesing JJ, et al. Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood. 2001;98(8):2466–73.
Neven B, et al. Defective anti-polysaccharide response and splenic marginal zone disorganization in ALPS patients. Blood. 2014;124(10):1597–609.
Bleesing JJ. Autoimmune lymphoproliferative syndrome (ALPS). Curr Pharm Des. 2003;9(3):265–78.
Hsu AP, et al. Autoimmune lymphoproliferative syndrome due to FAS mutations outside the signal-transducing death domain: molecular mechanisms and clinical penetrance. Genet Med. 2012;14(1):81–9.
Lambotte O, et al. Diagnosis of autoimmune lymphoproliferative syndrome caused by FAS deficiency in adults. Haematologica. 2013;98(3):389–92.
Ahn TS, et al. Whole exome sequencing reveals pathogenic variants in ADA2 and FAS causing DADA2 and ALPS. J Clin Immunol. 2023;43(6):1147–51.
Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–7.
Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–6.
Oliveira JB, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104(21):8953–8.
Niemela JE, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117(10):2883–6.
Patiroglu T, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol. 2015;159(1):58–62.
Lopez-Nevado M, et al. Primary Immune Regulatory disorders with an Autoimmune Lymphoproliferative Syndrome-Like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol. 2021;12:671755.
Infante AJ, et al. The clinical spectrum in a large kindred with autoimmune lymphoproliferative syndrome caused by a Fas mutation that impairs lymphocyte apoptosis. J Pediatr. 1998;133(5):629–33.
Bleesing JJH, Zhang NC. Autoimmune lymphoproliferative syndrome. In: Adam FJ, Mirzaa MP GM, et al. editors. GeneReviews® [Internet]. Editor: University of Washington: Seattle (WA); 2017.
Dowdell KC, et al. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood. 2010;115(25):5164–9.
Jackson CE, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64(4):1002–14.
Roesler J, et al. Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95DeltaTM), is the basis for ALPS Ia in a family with duplicated 3’ splice site AG in CD95 intron 5 on one allele. Blood. 2005;106(5):1652–9.
Corrionero A, et al. Strict 3’ splice site sequence requirements for U2 snRNP recruitment after U2AF binding underlie a genetic defect leading to autoimmune disease. RNA. 2011;17(3):401–11.
Fuchs H, et al. Residual CD95-pathway function in children with autoimmune lymphoproliferative syndrome is independent from clinical state and genotype of CD95 mutation. Pediatr Res. 2009;65(2):163–8.
Hadjadj J, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. 2019;134(1):9–21.
Agrebi N, et al. Rare splicing defects of FAS underly severe recessive autoimmune lymphoproliferative syndrome. Clin Immunol. 2017;183:17–23.
Tessarin G, et al. Complete CD95/FAS deficiency due to complex homozygous germline TNFRSF6 mutations in an adult patient with mild autoimmune lymphoproliferative syndrome (ALPS). Clin Immunol. 2021;228:108757.
Magerus-Chatinet A, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 2011;121(1):106–12.
Lopez-Nevado M, et al. Next generation sequencing for detecting somatic FAS mutations in patients with autoimmune lymphoproliferative syndrome. Front Immunol. 2021;12:656356.
Heurtier L, et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause activated PI3K-delta syndrome 1 (APDS1). Haematologica. 2017;102(7):e278–81.
Luo Y, et al. Identification of a novel de novo gain-of-function mutation of PIK3CD in a patient with activated phosphoinositide 3-kinase delta syndrome. Clin Immunol. 2018;197:60–7.
Elkaim E, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immunol. 2016;138(1):210–e2189.
Moreno-Corona N, et al. Two Monogenetic disorders, activated PI3-Kinase-delta syndrome 2 and Smith-Magenis Syndrome, in one patient: Case Report and a literature review of neurodevelopmental impact in primary immunodeficiencies Associated with disturbed PI3K signaling. Front Pediatr. 2021;9:688022.
Andrews A, et al. Genetic characterization of short stature patients with overlapping features of growth hormone insensitivity syndromes. J Clin Endocrinol Metab. 2021;106(11):e4716–33.
Molnar E, et al. Key diagnostic markers for autoimmune lymphoproliferative syndrome with molecular genetic diagnosis. Blood. 2020;136(17):1933–45.
Rensing-Ehl A, et al. Abnormally differentiated CD4 + or CD8 + T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood. 2014;124(6):851–60.
Maccari ME et al. A distinct CD38 + CD45RA + population of CD4+, CD8+, and double-negative T cells is controlled by FAS. J Exp Med, 2021. 218(2).
Kuehn HS, et al. FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol. 2011;186(10):6035–43.
Similuk MN, et al. Clinical exome sequencing of 1000 families with complex immune phenotypes: toward comprehensive genomic evaluations. J Allergy Clin Immunol. 2022;150(4):947–54.
Ben-Mustapha I, Agrebi N, Barbouche MR. Novel insights into FAS defects underlying autoimmune lymphoproliferative syndrome revealed by studies in consanguineous patients. J Leukoc Biol. 2018;103(3):501–8.
Neven B, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011;118(18):4798–807.
Buratti E, et al. Aberrant 5’ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35(13):4250–63.
Zhang MQ. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7(5):919–32.
Rieux-Laucat F, et al. Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations. Blood. 1999;94(8):2575–82.
Thaventhiran JED, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583(7814):90–5.
van den Berg A, et al. Germline FAS gene mutation in a case of ALPS and NLP Hodgkin lymphoma. Blood. 2002;99(4):1492–4.
Vaishnaw AK, et al. The molecular basis for apoptotic defects in patients with CD95 (Fas/Apo-1) mutations. J Clin Invest. 1999;103(3):355–63.
Richards DG, Stevenson GW. Laxatives prior to small bowel follow-through: are they necessary for a rapid and good-quality examination? Gastrointest Radiol. 1990;15(1):66–8.
van den Berg A, et al. FAS gene mutation in a case of autoimmune lymphoproliferative syndrome type IA with accumulation of gammadelta + T cells. Am J Surg Pathol. 2003;27(4):546–53.
Wang L, et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17(11):1324–9.
Arico M, et al. Variations of the UNC13D gene in patients with autoimmune lymphoproliferative syndrome. PLoS ONE. 2013;8(7):e68045.
Aydin Koker S, et al. Letter to the editor: coexistence of Autoimmune Lymphoproliferative Syndrome and Familial Mediterranean Fever. Iran J Immunol. 2020;17(2):172–4.
Hashem H, et al. Successful reduced intensity hematopoietic cell transplant in a patient with deficiency of adenosine deaminase 2. Bone Marrow Transpl. 2017;52(11):1575–6.
Hashem H, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood. 2017;130(24):2682–8.
Ulirsch JC, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet. 2018;103(6):930–47.
Carmona-Rivera C, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood. 2019;134(4):395–406.
Ozen S, et al. A monogenic disease with a Variety of Phenotypes: Deficiency of Adenosine Deaminase 2. J Rheumatol. 2020;47(1):117–25.
Schwab C, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–46.
Rojas-Restrepo J, et al. Establishing the Molecular diagnoses in a cohort of 291 patients with predominantly antibody Deficiency by targeted next-generation sequencing: experience from a Monocentric Study. Front Immunol. 2021;12:786516.
Besnard C, et al. Pediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol. 2018;188:52–7.
Ayrignac X et al. Two neurologic facets of CTLA4-related haploinsufficiency. Neurol Neuroimmunol Neuroinflamm, 2020. 7(4).
Stray-Pedersen A, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232–45.
Hoyos-Bachiloglu R, et al. The many faces of XMEN Disease, Report of two patients with novel mutations. J Clin Immunol. 2020;40(2):415–7.
Gadoury-Levesque V, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. 2020;4(12):2578–94.
De Filippi P, et al. Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol. 2009;147(5):706–9.
Altmuller F, et al. Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet. 2017;25(7):823–31.
Kiel C, Serrano L. Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations. Mol Syst Biol. 2014;10(5):727.
Wang W, et al. RAS-associated autoimmune leukoproliferative disease (RALD) manifested with early-onset SLE-like syndrome: a case series of RALD in Chinese children. Pediatr Rheumatol Online J. 2019;17(1):55.
Acknowledgements
We thank the ordering providers that referred patients to our institution. We also thank the molecular genetic laboratory staff and the diagnostic immunology laboratory staff at CCHMC for offering and performing the assays.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Conceptualization and design: XX, JB, and WZ. Data collection: XX, JD, WZ. ALPS genetic reporting review: YW, JL, QG, DBD, WZ. ALPS immunology test and apoptosis test reporting review: JB. Data analysis and manuscript drafting: XX, JB, and WZ. All authors reviewed and contributed to the manuscript. Study supervision: JB and WZ.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Denton, J., Wu, Y. et al. Genetic Testing in Patients with Autoimmune Lymphoproliferative Syndrome: Experience of 802 Patients at Cincinnati Children’s Hospital Medical Center. J Clin Immunol 44, 166 (2024). https://doi.org/10.1007/s10875-024-01772-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10875-024-01772-z