
Abstract
Molecular diagnosis of inborn errors of immunity (IEI) plays a critical role in determining patients’ long-term prognosis, treatment options, and genetic counseling. Over the past decade, the broader utilization of next-generation sequencing (NGS) techniques in both research and clinical settings has facilitated the evaluation of a significant proportion of patients for gene variants associated with IEI. In addition to its role in diagnosing known gene defects, the application of high-throughput techniques such as targeted, exome, and genome sequencing has led to the identification of novel disease-causing genes. However, the results obtained from these different methods can vary depending on disease phenotypes or patient characteristics. In this study, we conducted whole-exome sequencing (WES) in a sizable cohort of IEI patients, consisting of 303 individuals from 21 different clinical immunology centers in Türkiye. Our analysis resulted in likely genetic diagnoses for 41.1% of the patients (122 out of 297), revealing 52 novel variants and uncovering potential new IEI genes in six patients. The significance of understanding outcomes across various IEI cohorts cannot be overstated, and we believe that our findings will make a valuable contribution to the existing literature and foster collaborative research between clinicians and basic science researchers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inborn errors of immunity or primary immunodeficiencies (PIDs) represent a diverse group of disorders characterized by increased susceptibility to infections, malignancy, allergy, and immune dysregulation [1]. While these diseases occur at a frequency of approximately 1 in 10,000 in the general population, their prevalence is higher in societies with elevated rates of consanguinity, such as Türkiye [2,3,4]. The genetic pleiotropy and heterogeneity observed in IEI contribute to the broad range of clinical manifestations associated with these disorders [5]. The majority of IEI cases are monogenic diseases with autosomal recessive inheritance patterns [5]. Therefore, comprehensive genetic diagnosis is vital for effective management of patients with IEI. In the past decade, NGS methods have revolutionized genetic screening, greatly enhancing the diagnostic capabilities for IEI [6]. This progress has led to an unprecedented increase in the identification of genes causing immunodeficiencies, with approximately 500 genetic defects associated with immunodeficiency currently recognized [7].
Founded in 2018 in memory of Can Sucak, who suffered from ZAP70 deficiency, the Candan Bişeyler Foundation (CSCBF) actively supports research in the field of IEI and raises awareness in Türkiye. The “Hacettepe University Can Sucak Research Laboratory for Translational Immunology” is dedicated to providing genetic diagnosis for immunodeficiency patients and conducting advanced functional research in a comprehensive manner throughout the country. This study presents the results of a comprehensive investigation into the genetic diagnosis of an extensive cohort of IEI patients from a specialized immune deficiency research center in Türkiye.
Methods
Study Participants
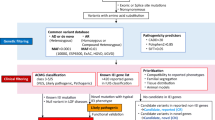
Patients diagnosed with IEI based on clinical and laboratory characteristics between 2020 and 2023 were included in the study. These patients were recruited from multiple clinical immunology centers in Türkiye. Blood samples were collected from the patients following the guidelines and approval of the local Ethics Committee of Hacettepe University. Informed consent forms were obtained from the participants or their parents. The study's workflow is illustrated in Fig. 1.

Schematic workflow of the study
Whole Exome Sequencing and Variant Analysis
Genomic DNA was isolated from peripheral blood samples using a DNA isolation kit (GeneAll). The NGS exome library was prepared utilizing the Illumina Nextera DNA Prep with Enrichment Kit. Sequencing was carried out on the Illumina NextSeq 550 platform, generating 150-bp paired-end reads. Mapping, variant calling, and annotation were performed using SEQ Platform v8 (Genomize). Copy number variation (CNV) analysis was conducted using SEQ Platform as well.
To identify causative variants, we employed a filtering strategy that involved screening all variants identified from the WES data. Our focus was on exonic and splice site variants, excluding synonymous variants, and we specifically looked for rare variants with a minor allele frequency of less than 1% in different strategic gene groups. Initially, we examined rare variants in known IEI genes (approximately 500), followed by potential candidate genes predicted by the human gene connectome [8]. Finally, we assessed variants across the entire set of genes (Supplementary Figure 2A).
Sanger Sequencing
To validate the identified variants, we conducted Sanger sequencing using standard protocols [9].
RT-qPCR
RT-qPCR was utilized to validate the effects of structural variants. Total RNA was isolated from peripheral blood mononuclear cells (PBMCs) obtained from both patients and healthy controls using the NucleoSpin RNA Plus Kit (Macherey-Nagel). Subsequently, cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was carried out on the CFX Connect System (Bio-Rad) using the iTaq Universal SYBR Green Supermix (Bio-Rad) [10].
Results
Technical Output of the Sequencing Data
The results of the WES data showed a total number of reads ranging from 21.7 to 77.6 million (median: 46.1) (Supplementary Figure 2B). The average depth of coverage varied between 24.5 and 134.2 (median: 64.1) (Supplementary Figure 2C). The target regions (exons and splice regions) were covered at a depth of 20X from 89.02% to 99.91%, and at a depth of 50X from 68.13% to 99.65% (Supplementary Figure 2D).
Patients
Our study involved a total of 303 individuals who were clinically diagnosed with IEI. These participants were recruited from 21 separate clinical immunology centers and they were selected after assessments with their clinicians. Especially, patients truly exhibited severe phenotypes of immunodeficiency were admitted to the study. However, six patients were excluded from the current analysis as they exhibited potential novel IEI-associated genes, pending further investigation through functional studies. Therefore, the analysis in this study includes 297 patients.
Among the included patients, there were 145 males and 152 females, representing a relatively balanced gender distribution. The age range of the participants varied from three months to 42 years, with a median age of nine years. The majority of the cohort consisted of pediatric patients (n=252), while a smaller subset comprised adult patients (n=45). A notable observation in our study was the high consanguinity rate, with 64.6% (192 out of 297 cases) of patients demonstrating consanguineous relationships within their families. The distribution of clinical diagnoses, classified according to the International Union of Immunological Societies (IUIS) classification, included 27 cases of Severe Combined Immunodeficiency (SCID), 105 cases of Combined Immunodeficiency (CID), 64 cases of Primary Antibody Deficiency (PAD), 49 cases of Primary Immune Regulatory Disorder (PIRD), 22 cases of congenital anomalies affecting phagocyte number/function, 17 cases of disorders of intrinsic and innate immunity, 10 cases of autoinflammatory disorders, and 3 cases of other classified IEI. These other cases potentially involve bone marrow failure or complement deficiencies, as illustrated in Fig. 2A.

Patient and variant characteristics. A Distribution of the patients based on their clinical diagnosis. B Diagnostic yield of the patients. C Number of the detected variants and their distribution across different IEI genes. D Types of detected variants and their novelty. E Distribution of zygosity. F Number of diagnosis in patient groups
Results of Genetic Diagnosis and the Profile of Disease-Causing Variants
In our cohort, a genetic diagnosis was established in 122 out of the 297 patients examined, with a total of 127 potential genetic variants identified. This yielded a diagnostic rate of 41.1%. Among the 193 patients with consanguineous parents, causative genetic defects were identified in 95 individuals, resulting in a diagnostic rate of 49.7%. On the other hand, among the 106 patients from non-consanguineous parents, 28 individuals (25.7%) received a genetic diagnosis. The diagnostic rate was higher in pediatric patients, with 44.4% (112 out of 252) receiving a genetic diagnosis, compared to the adult group, which had a lower rate of 22% (10 out of 45) (Fig. 2B). Details of all identified genetic variants and their associated clinical features are presented in Table 1, Table 2 and Supplementary Table 3. In addition, variant characteristics including American College of Medical Genetics (ACMG) criteria and pathogenicity prediction scores were given in Supplementary Table 1). Overall, a total of 127 likely causative genetic anomalies were identified across 64 known IEI genes, as depicted in Fig. 2C. Among these genetic variants, 75 had been previously reported in public databases, while 52 were novel findings reported in this study (Fig. 2D). The variants consisted of 92 homozygous, 27 heterozygous, and 8 hemizygous mutations (Fig. 2E). The spectrum of variant types included 69 missense mutations, 24 nonsense mutations, 22 insertion/deletions (indels), 9 essential splice site variations, and 3 copy number variations (Figure 2D). CNV analysis was performed on 57 subjects using a strategy that incorporated samples with comparable mean read depths. The implications of the CNVs were validated through capillary sequencing or quantitative PCR (qPCR). The causality of monoallelic variants was evaluated based on clinical and laboratory features of the patients, literature associations, or different functional analyses (Supplementary Table 2). The diagnostic rates across different disease categories were as follows: Severe Combined Immunodeficiency (SCID) had a diagnostic rate of 100%, congenital anomalies affecting phagocyte number/function at 68.1%, autoinflammatory disorders at 50%, Primary Immune Regulatory Disorder (PIRD) at 46.9%, intrinsic and innate immunity defects at 41.1%, Combined Immunodeficiency (CID) at 32.3%, other forms of IEI at 33.3%, and Primary Antibody Deficiency (PAD) at 15.6%, and (Fig. 2F).
Discussion
Advancements in NGS, with WES at the forefront, have been instrumental in the diagnostic processes of IEI by pinpointing causative genetic aberrations [72]. Genetic diagnosis now routinely assists in the delineation of IEI, underscoring its significance in the strategic management of patient treatments. Literature suggests a wide-ranging diagnostic yield for targeted and exome sequencing, from 10% to 70%, across various IEI patient groups [23, 58, 68, 73,74,75,76,77,78,79] . In this study, out of the 127 causative genetic defects in 122 patients, we identified 52 novel IEI-causing variants. We also discovered novel and very rare gene variants in NFATC2, CHUK, and PIK3CG genes, which have limited reported cases in the literature [80,81,82,83].
Among the 297 patients evaluated, a genetic etiology was confirmed in 122 individuals, resulting in a diagnostic yield of 41.1%. Diagnostic success exhibited pronounced variation among the different IEI subtypes: cases of SCID reached a 100% genetic identification rate, whereas CID and PID manifested lower diagnostic rates of 31% and 45%, respectively. Within the PAD cohort, genetic causality was determined in a mere 15.6% of cases (10 patients). This notably diminished diagnostic yield in Primary Antibody Deficiencies is in concordance with prior regional studies conducted by Fırtına S et al. [84]. In contrast, patients with probable Mendelian susceptibility to mycobacterial diseases and chronic granulomatous disease (CGD) demonstrated significantly higher diagnostic rates, with near-complete success in CGD patients.
The discrepancies in diagnostic success among IEI subtypes are primarily attributed to the complex nature of these disorders rather than limitations of WES. Factors such as the specific type of immunodeficiency, diverse clinical presentations, patient medical histories, and environmental influences affect the probability of achieving a genetic diagnosis [72]. Other factors include variable gene penetrance, the distinction between monogenic and polygenic influences, and various environmental considerations such as pathogenic exposures and age at presentation [85, 86]. Consanguinity plays a significant role in genetic diagnosis, as most IEI cases have autosomal recessive inheritance. Consanguineous populations or those from isolated regions with distinct phenotypes have reported higher diagnostic yields [87]. In our study, the consanguinity rate was 64.6%, and a diagnosis was made in 49.7% of those cases. We found 27 heterozygous variants in 21 unrelated patients, which can provide insights into the impact of heterozygous variants on protein function and aid in the search for novel IEI genes.
Currently, approximately 500 genetic etiologies leading to IEI are known [7]. Although the use of NGS, particularly WES, is increasing, it has limitations. Exome sequencing focuses on coding regions and essential splice sites, making it challenging to detect structural variations [72] and the use of short-read sequencing as in our study makes it difficult to map reads to repeated sequences, and pseudogenes [88]. Long-read sequencing (LRS) technologies both for exome or genome, have the capacity to enhance the detection of genetic variations and regions that are challenging to analyze with existing short-read NGS techniques [88,89,90]. However, the cost and complexity of analyzing large datasets pose challenges for WGS. In our study, we only identified three structural variants in 57 patients. Nevertheless, studies have shown the effectiveness of WGS in detecting both CNVs and coding variants [91, 92]. Reducing the cost of WGS and developing user-friendly bioinformatic tools may make it a routine diagnostic approach for IEI screening.
In conclusion, our findings highlight the limited success of WES in the genetic investigation of presumed IEI. The prospective adoption of WGS could enhance diagnostic yields, potentially surpassing WES in clinical examinations. With our substantial study cohort and diverse clinical presentations, the genetic variations we have identified will significantly contribute to the diagnosis of future IEI cases and guide the development of optimized NGS panels for these conditions.
Data Availability
No datasets were generated or analysed during the current study.
References
Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human inborn errors of immunity: An expanding universe. Sci Immunol. 2020;5(49)
Eldeniz FC, Gul Y, Yorulmaz A, Guner SN, Keles S, Reisli I. Evaluation of the 10 Warning Signs in Primary and Secondary Immunodeficient Patients. Front Immunol. 2022;13:900055.
Meyts I, Bousfiha A, Duff C, Singh S, Lau YL, Condino-Neto A, et al. Primary Immunodeficiencies: A Decade of Progress and a Promising Future. Front Immunol. 2020;11:625753.
Sanal O, Tezcan I. Thirty years of primary immunodeficiencies in Turkey. Ann N Y Acad Sci. 2011;1238:15–23.
Conley ME, Casanova JL. Discovery of single-gene inborn errors of immunity by next generation sequencing. Curr Opin Immunol. 2014;30:17–23.
Vorsteveld EE, Hoischen A, van der Made CI. Next-Generation Sequencing in the Field of Primary Immunodeficiencies: Current Yield, Challenges, and Future Perspectives. Clin Rev Allergy Immunol. 2021;61(2):212–25.
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–507.
Itan Y, Casanova JL. Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Front Immunol. 2015;6:142.
Halacli SO, Ayvaz DC, Sun-Tan C, Erman B, Uz E, Yilmaz DY, et al. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: A novel mutation. Clin Immunol. 2015;161(2):316–23.
Aba Ü, Maslak IC, Ipsir C, Pehlivan D, Warnock NI, Tumes DJ, et al. A Novel Homozygous Germline Mutation in Transferrin Receptor 1 (TfR1) Leads to Combined Immunodeficiency and Provides New Insights into Iron-Immunity Axis. J Clin Immunol. 2024;44(2)
Erman B, Firtina S, Aksoy BA, Aydogdu S, Genc GE, Dogan O, et al. Invasive Saprochaete capitata Infection in a Patient with Autosomal Recessive CARD9 Deficiency and a Review of the Literature. J Clin Immunol. 2020;40(3):466–74.
Erman B, Cipe F. Genetic Screening of the Patients with Primary Immunodeficiency by Whole-Exome Sequencing. Pediatr Allergy Immunol Pulmonol. 2020;33(1):19–24.
Erman B, Firtina S, Fisgin T, Bozkurt C, Cipe FE. Biallelic Form of a Known CD3E Mutation in a Patient with Severe Combined Immunodeficiency. J Clin Immunol. 2020;40(3):539–42.
Ghosh S, Kostel Bal S, Edwards ESJ, Pillay B, Jimenez Heredia R, Erol Cipe F, et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood. 2020;136(23):2638–55.
Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24)
Meshaal SS, El Hawary RE, Abd Elaziz DS, Eldash A, Alkady R, Lotfy S, et al. Phenotypical heterogeneity in RAG-deficient patients from a highly consanguineous population. Clin Exp Immunol. 2019;195(2):202–12.
Tabori U, Mark Z, Amariglio N, Etzioni A, Golan H, Biloray B, et al. Detection of RAG mutations and prenatal diagnosis in families presenting with either T-B severe combined immunodeficiency or Omenn's syndrome. Clinical Genetics. 2004;65(4):322–6.
Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, Geha RS. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37(8):829–34.
Salzer U, Chapel HM, Webster ADB, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37(8):820–8.
Köker MY, van Leeuwen K, de Boer M, Çelmeli F, Metin A, Özgür TT, et al. Six different mutations including three novel mutations in ten families from Turkey, resulting in autosomal recessive chronic granulomatous disease. Eur J Clin Invest. 2009;39(4):311–9.
Rae J, Noack D, Heyworth PG, Ellis BA, Curnutte JT, Cross AR. Molecular analysis of 9 new families with chronic granulomatous disease caused by mutations in, the gene encoding p22. Blood. 2000;96(3):1106–12.
Sefer AP, Abolhassani H, Ober F, Kayaoglu B, Bilgic Eltan S, Kara A, et al. Expanding the Clinical and Immunological Phenotypes and Natural History of MALT1 Deficiency. J Clin Immunol. 2022;42(3):634–52.
Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. 2018;93(3):647–55.
Egg D, Rump IC, Mitsuiki N, Rojas-Restrepo J, Maccari ME, Schwab C, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol. 2022;149(2):736–46.
Hoshino A, Tanita K, Kanda K, Imadome KI, Shikama Y, Yasumi T, et al. High frequencies of asymptomatic Epstein-Barr virus viremia in affected and unaffected individuals with CTLA4 mutations. Clin Immunol. 2018;195:45–8.
Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–46.
Minegishi Y, Coustan-Smith E, Wang YH, Cooper MD, Campana D, Conley ME. Mutations in the human λ5/14.1 gene result in B cell deficiency and agammaglobulinemia. J Exp Med. 1998;187(1):71–7.
Kulkarni M, Hule G, de Boer M, van Leeuwen K, Kambli P, Aluri J, et al. Approach to Molecular Diagnosis of Chronic Granulomatous Disease (CGD): an Experience from a Large Cohort of 90 Indian Patients. J Clin Immunol. 2018;38(8):898–916.
Noack D, Rae J, Cross AR, Muñoz J, Salmen S, Mendoza JA, et al. Autosomal recessive chronic granulomatous disease caused by novel mutations in , the gene encoding the p67-component of phagocyte NADPH oxidase. Hum Genet. 1999;105(5):460–7.
de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 2010;89(6):381–402.
Hatipoglu N, Guvenc BH, Deswarte C, Koksalan K, Boisson-Dupuis S, Casanova JL, Bustamante J. Inherited IL-12Rbeta1 Deficiency in a Child With BCG Adenitis and Oral Candidiasis: A Case Report. Pediatrics. 2017;140(5)
Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O'Neil LJ, Liu Y, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood. 2019;134(4):395–406.
Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood. 2017;130(24):2682–8.
Hashem H, Vatsayan A, Gupta A, Nagle K, Hershfield M, Dalal J. Successful reduced intensity hematopoietic cell transplant in a patient with deficiency of adenosine deaminase 2. Bone Marrow Transplant. 2017;52(11):1575–6.
Asilsoy S, Bilgili G, Turul T, Dizdarer C, Kalkan S, Yasli H, et al. Interleukin-12/-23 receptor beta 1 deficiency in an infant with draining BCG lymphadenitis. Pediatr Int. 2009;51(2):310–2.
Fieschi C, Dupuis S, Catherinot E, Feinberg J, Bustamante J, Breiman A, et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications. J Exp Med. 2003;197(4):527–35.
Park AY, Leney-Greene M, Lynberg M, Gabrielski JQ, Xu X, Schwarz B, et al. GIMAP5 deficiency reveals a mammalian ceramide-driven longevity assurance pathway. Nat Immunol. 2024;25(2):282–93.
Bettinardi A, Brugnoni D, Quiros-Roldan E, Malagoli A, La Grutta S, Correra A, Notarangelo LD. Missense mutations in the Fas gene resulting in autoimmune lymphoproliferative syndrome: a molecular and immunological analysis. Blood. 1997;89(3):902–9.
Balta G, Okur H, Unal S, Yarali N, Gunes AM, Unal S, et al. Assessment of clinical and laboratory presentations of familial hemophagocytic lymphohistiocytosis patients with homozygous W374X mutation. Leuk Res. 2010;34(8):1012–7.
Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, Hennies HC. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27(1):62–8.
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–7.
Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111(10):4954–7.
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39(1):86–92.
Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. 2011;43(2):127–31.
Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95(1):96–107.
Rowe JH, Delmonte OM, Keles S, Stadinski BD, Dobbs AK, Henderson LA, et al. Patients with CD3G mutations reveal a role for human CD3gamma in T(reg) diversity and suppressive function. Blood. 2018;131(21):2335–44.
Teimourian S, Zomorodian E, Badalzadeh M, Pouya A, Kannengiesser C, Mansouri D, et al. Characterization of six novel mutations in CYBA: the gene causing autosomal recessive chronic granulomatous disease. Br J Haematol. 2008;141(6):848–51.
Cetica V, Hackmann Y, Grieve S, Sieni E, Ciambotti B, Coniglio ML, et al. Patients with Griscelli syndrome and normal pigmentation identify RAB27A mutations that selectively disrupt MUNC13-4 binding. J Allergy Clin Immunol. 2015;135(5):1310–8 e1.
Mamishi S, Modarressi MH, Pourakbari B, Tamizifar B, Mahjoub F, Fahimzad A, et al. Analysis of RAB27A gene in griscelli syndrome type 2: novel mutations including a deletion hotspot. J Clin Immunol. 2008;28(4):384–9.
Sarper N, Ipek IO, Ceran O, Karaman S, Bozaykut A, Inan S. A rare syndrome in the differential diagnosis of hepatosplenomegaly and pancytopenia: report of identical twins with Griscelli disease. Ann Trop Paediatr. 2003;23(1):69–73.
Sepulveda FE, Debeurme F, Menasche G, Kurowska M, Cote M, Pachlopnik Schmid J, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood. 2013;121(4):595–603.
Cagdas D, Halacli SO, Tan C, Lo B, Cetinkaya PG, Esenboga S, et al. A Spectrum of Clinical Findings from ALPS to CVID: Several Novel LRBA Defects. J Clin Immunol. 2019;39(7):726–38.
Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223–30.
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–40.
Esenboga S, Akal C, Karaatmaca B, Erman B, Dogan S, Orhan D, et al. Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature. Clin Immunol. 2018;197:1–5.
Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J Allergy Clin Immunol. 2015;135(6):1578–88 e5.
van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119(1):91–8.
Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. 2016;138(4):1142–51 e2.
Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125(11):4135–48.
Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J Allergy Clin Immunol. 2017;139(2):597–606 e4.
Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88–97.
Altare F, Ensser A, Breiman A, Reichenbach J, Baghdadi JE, Fischer A, et al. Interleukin-12 receptor beta1 deficiency in a patient with abdominal tuberculosis. J Infect Dis. 2001;184(2):231–6.
Sakai T, Matsuoka M, Aoki M, Nosaka K, Mitsuya H. Missense mutation of the interleukin-12 receptor beta1 chain-encoding gene is associated with impaired immunity against Mycobacterium avium complex infection. Blood. 2001;97(9):2688–94.
Adams SP, Wilson M, Harb E, Fairbanks L, Xu-Bayford J, Brown L, et al. Spectrum of mutations in a cohort of UK patients with ADA deficient SCID: Segregation of genotypes with specific ethnicities. Clin Immunol. 2015;161(2):174–9.
Lee PP, Chan KW, Chen TX, Jiang LP, Wang XC, Zeng HS, et al. Molecular diagnosis of severe combined immunodeficiency--identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. J Clin Immunol. 2011;31(2):281–96.
Reiff A, Bassuk AG, Church JA, Campbell E, Bing X, Ferguson PJ. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J Clin Immunol. 2013;33(8):1289–92.
Zhang ZY, Zhao XD, Jiang LP, Liu EM, Cui YX, Wang M, et al. Clinical characteristics and molecular analysis of three Chinese children with Omenn syndrome. Pediatr Allergy Immunol. 2011;22(5):482–7.
Simon AJ, Golan AC, Lev A, Stauber T, Barel O, Somekh I, et al. Whole exome sequencing (WES) approach for diagnosing primary immunodeficiencies (PIDs) in a highly consanguineous community. Clin Immunol. 2020;214:108376.
Niemela J, Kuehn HS, Kelly C, Zhang M, Davies J, Melendez J, et al. Caspase-8 Deficiency Presenting as Late-Onset Multi-Organ Lymphocytic Infiltration with Granulomas in two Adult Siblings. J Clin Immunol. 2015;35(4):348–55.
Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358(19):2030–8.
Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93(5):885–96.
Picard C, Fischer A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol. 2014;44(10):2854–61.
Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. 2016;137(6):1780–7.
Bisgin A, Boga I, Yilmaz M, Bingol G, Altintas D. The Utility of Next-Generation Sequencing for Primary Immunodeficiency Disorders: Experience from a Clinical Diagnostic Laboratory. Biomed Res Int. 2018;2018:9647253.
Erman B, Bilic I, Hirschmugl T, Salzer E, Boztug H, Sanal O, et al. Investigation of Genetic Defects in Severe Combined Immunodeficiency Patients from Turkey by Targeted Sequencing. Scand J Immunol. 2017;85(3):227–34.
Kojima D, Wang X, Muramatsu H, Okuno Y, Nishio N, Hama A, et al. Application of extensively targeted next-generation sequencing for the diagnosis of primary immunodeficiencies. J Allergy Clin Immunol. 2016;138(1):303–5 e3.
Moens LN, Falk-Sorqvist E, Asplund AC, Bernatowska E, Smith CI, Nilsson M. Diagnostics of primary immunodeficiency diseases: a sequencing capture approach. PLoS One. 2014;9(12):e114901.
Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133(2):529–34.
Okano T, Imai K, Naruto T, Okada S, Yamashita M, Yeh TW, et al. Whole-Exome Sequencing-Based Approach for Germline Mutations in Patients with Inborn Errors of Immunity. J Clin Immunol. 2020;40(5):729–40.
Bainter W, Lougaris V, Wallace JG, Badran Y, Hoyos-Bachiloglu R, Peters Z, et al. Combined immunodeficiency with autoimmunity caused by a homozygous missense mutation in inhibitor of nuclear factor ?B kinase alpha (IKKalpha). Sci Immunol. 2021;6(63):eabf6723.
Sharma M, Fu MP, Lu HY, Sharma AA, Modi BP, Michalski C, et al. Human complete NFAT1 deficiency causes a triad of joint contractures, osteochondromas, and B-cell malignancy. Blood. 2022;140(17):1858–74.
Takeda AJ, Maher TJ, Zhang Y, Lanahan SM, Bucklin ML, Compton SR, et al. Human PI3Kgamma deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat Commun. 2019;10(1):4364.
Thian M, Hoeger B, Kamnev A, Poyer F, Kostel Bal S, Caldera M, et al. Germline biallelic PIK3CG mutations in a multifaceted immunodeficiency with immune dysregulation. Haematologica. 2020;105(10):e488.
Firtina S, Ng YY, Ng OH, Kiykim A, Ozek EY, Kara M, et al. Primary antibody deficiencies in Turkey: molecular and clinical aspects. Immunol Res. 2022;70(1):44–55.
Edwards ESJ, Bosco JJ, Ojaimi S, O'Hehir RE, van Zelm MC. Beyond monogenetic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency. Cell Mol Immunol. 2021;18(3):588–603.
Rojas-Restrepo J, Caballero-Oteyza A, Huebscher K, Haberstroh H, Fliegauf M, Keller B, et al. Establishing the Molecular Diagnoses in a Cohort of 291 Patients With Predominantly Antibody Deficiency by Targeted Next-Generation Sequencing: Experience From a Monocentric Study. Front Immunol. 2021;12:786516.
Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. 2018;141(4):1450–8.
Mantere T, Kersten S, Hoischen A. Long-Read Sequencing Emerging in Medical Genetics. Front Genet. 2019;10:426.
Sanford Kobayashi E, Batalov S, Wenger AM, Lambert C, Dhillon H, Hall RJ, et al. Approaches to long-read sequencing in a clinical setting to improve diagnostic rate. Sci Rep. 2022;12(1):16945.
Troskie RL, Jafrani Y, Mercer TR, Ewing AD, Faulkner GJ, Cheetham SW. Long-read cDNA sequencing identifies functional pseudogenes in the human transcriptome. Genome Biol. 2021;22(1):146.
Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112(17):5473–8.
Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583(7814):90–5.
Acknowledgements
We express our gratitude to the “Can Sucak Candan Biseyler” Foundation (CSCBF) for their valuable support and contributions throughout this study. The CSCBF was established in 2018 to honor the memory of Can Sucak, who tragically passed away due to complications of primary immunodeficiency. The foundation actively supports research in the field of primary immunodeficiency and raises awareness about this condition. Additionally, we would like to acknowledge The Hospital Research Foundation (THRF) for their support of GC.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). The study received support from the “Sucak Candan Biseyler” Foundation and the Clinical Immunology Society, which provided the necessary Whole Exome Sequencing (WES) kits for the research.
Author information
Authors and Affiliations
Contributions
B. E, U. A, C. I, D. P, B. O, C. B, S. T and M. K performed the experiments and analyzed data the with G. C. C. A, Ç. A, F. Ç, G. S, S. B. E, A. O, S. B, E. K. A, A. K, B. K, H. U, D. F. K, F. Ç, T. A, D. Ö, E. A, E. S. A, E. K, M. K, M. Y, Z. B, S. A, D.Ç.A, Ö. K, A. P. S, Ş. N. G, S. K, I. R, U. M, N. D. C, Ş. H, S. S. K, A. M, F. D, A. I and I. T provided clinical care of the patients, clinical data and patient materials. B. E, G. C, A. I and I. T wrote the manuscript. B. E, A. I, and I. T conceptualized and coordinated the study and provided laboratory resources. All authors critically reviewed the manuscript and agreed to its publication.
Corresponding author
Ethics declarations
Consent to Participate
Informed consent was obtained from all individual participants who were included in the study.
Consent for Publication
The manuscript does not contain any personal data of individual participants.
Conflict of Interests
The authors declare no competing interests.
Ethics Approval
This study was conducted in accordance with the principles outlined in the Declaration of Helsinki. Approval for the study was obtained from the local Ethics Committee of Hacettepe University (Approval number: GO 20/407).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 806 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Erman, B., Aba, U., Ipsir, C. et al. Genetic Evaluation of the Patients with Clinically Diagnosed Inborn Errors of Immunity by Whole Exome Sequencing: Results from a Specialized Research Center for Immunodeficiency in Türkiye. J Clin Immunol 44, 157 (2024). https://doi.org/10.1007/s10875-024-01759-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10875-024-01759-w