
Abstract
We report the updated classification of inborn errors of immunity, compiled by the International Union of Immunological Societies Expert Committee. This report documents the key clinical and laboratory features of 55 novel monogenic gene defects, and 1 phenocopy due to autoantibodies, that have either been discovered since the previous update (published January 2020) or were characterized earlier but have since been confirmed or expanded in subsequent studies. While variants in additional genes associated with immune diseases have been reported in the literature, this update includes only those that the committee assessed that reached the necessary threshold to represent novel inborn errors of immunity. There are now a total of 485 inborn errors of immunity. These advances in discovering the genetic causes of human immune diseases continue to significantly further our understanding of molecular, cellular, and immunological mechanisms of disease pathogenesis, thereby simultaneously enhancing immunological knowledge and improving patient diagnosis and management. This report is designed to serve as a resource for immunologists and geneticists pursuing the molecular diagnosis of individuals with heritable immunological disorders and for the scientific dissection of cellular and molecular mechanisms underlying monogenic and related human immune diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inborn errors of immunity (IEI) are caused by damaging germline variants in single genes. IEI present clinically as increased susceptibility to infections, autoimmunity, autoinflammatory diseases, allergy, bone marrow failure, and/or malignancy. While individually rare, the aggregated number of individuals with an IEI represents a significant health burden [1]. Genetic variants cause disease by altering the encoded gene product, such as by abolishing or reducing protein expression and function (null/hypomorphic) or modifying the protein to acquire gain-of-function (GOF) [2,3,4,5]. Mechanisms of disease in IEI depend on the nature of the variant as well as the mode of inheritance. Thus, monoallelic variants can cause disease by haploinsufficiency, negative dominance, or GOF. In contrast, biallelic genetic lesions (homozygous, compound heterozygous) cause autosomal recessive (AR) traits by loss of expression, loss of function (LOF), GOF, or even neomorphic function of the encoded protein, while X-linked recessive traits arise from LOF or GOF variants on the X chromosome, either in hemizygosity in males, or homozygous state in females.
The fact that some monogenic variants are pathogenic clearly highlights the non-redundant and fundamental roles of individual genes and proteins, and associated pathways and cell types, in the development and function of leukocytes and non-hematopoietic cells that contribute to immune homeostasis and host defense [6, 7]. Thus, IEI represent an elegant model linking defined monogenic defects with clinical phenotypes of immune dysregulation. IEI have also revealed mechanisms of disease pathogenesis in, and enabled the implementation of gene- or pathway-specific therapies for the treatment of, rare and common conditions and established fundamental aspects of human immunology [8,9,10]. Thus, the study of IEI has enabled profound advances in molecular medicine and human biology.
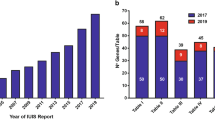
Since 1970, an international expert committee comprising pediatric and adult clinical immunologists, clinician/scientists and researchers in basic immunology — initially under the auspices of the World Health Organization and currently the International Union of Immunological Societies (IUIS) — has provided the clinical and research communities with an update of genetic causes of immune deficiency and dysregulation https://iuis.org/committees/iei/ (Fig. 1A).

Accumulative discovery of novel inborn errors of immunity: 1980–2022. (A) The number of genetic defects underlying monogenic immune disorders as reported in the indicated year. (B) The number of pathogenic variants listed in each Table of the IUIS IEI committee 2022 report. The numbers in each column correspond to the number of genes reported in the 2019 IUIS update (blue bars) [4, 5], the number of new genes for each Table contained in this report (red bars), and the total number of genes for each Table (black number). Note: The 14 conditions listed for Table 10 are either phenocopies of germline IEI due to somatic variants or neutralizing autoAbs. Somatic variants in UBA1 are also listed in Table 10, although there is currently no IEI resulting from germline UBA1 variants [97]
IEI are currently categorized into 10 Tables, with subtables segregating groups of disorders into overlapping phenotypes. These tables describe the following: combined immunodeficiencies (Table 1, 3 subtables); combined immunodeficiencies with syndromic features (Table 2; 9 subtables); predominantly antibody deficiencies (Table 3; 3 subtables); diseases of immune dysregulation (Table 4; 7 subtables); congenital defects of phagocytes (Table 5; 4 subtables); defects in intrinsic and innate immunity (Table 6; 9 subtables); autoinflammatory diseases (Table 7; 3 subtables); complement deficiencies (Table 8); bone marrow failure (Table 9), and phenocopies of inborn errors of immunity (Table 10) (Fig. 1B) [5].
The committee strives to publish an updated report approximately every 2 years to consolidate advances and catalog current IEIs (Fig. 1A) [5]. While COVID-19 has delayed producing this report in the desired timeframe, it has also uncovered several new IEI — some of these are highlighted below. Many genetic variants related to IEI have been reported recently. Rather than including every candidate gene reported in the peer-reviewed scientific literature, the committee applies stringent criteria to classify gene defects as novel causes of IEI [11]. These criteria include:
-
1.
The patient’s candidate genotype is monogenic and does not occur in individuals without the clinical phenotype (acknowledging that some conditions have incomplete penetrance).
-
2.
Experimental studies establish that the genetic variant impairs, destroys, or alters expression or function of the gene product.
-
3.
The causal relationship between the candidate genotype and the clinical phenotype must be confirmed via a relevant cellular phenotype, including — where possible — rescue of a functional defect [11].
These criteria can be met by publication of multiple cases from unrelated kindreds, including detailed immunologic data, or publication of very few — even single — cases for whom compelling mechanistic data are provided, often revealed from complementary studies in animal or cell culture models. We also considered whether sufficient justification was provided to exclude alternative candidate gene variants identified in single cases, the depth of the clinical descriptions of affected individuals, and the level of immune and mechanistic characterization. This 2022 update and the accompanying “Phenotypical IUIS Classification” publications are intended as resources for clinicians and researchers, as well as guiding the design of panels used for targeted gene sequencing to facilitate genetic diagnoses of IEI. Here, we summarize data on the genetic cause of 55 novel IEI, and 1 phenocopy due to autoantibodies, that have been assessed since the previous update [5] (Supplementary Table 1). Remarkably, 15 of the 55 novel IEI have come from the identification and extensive work-up of single patients. Two themes that are expanded in this new set of genes are narrow infection susceptibility and immune dysregulation, which collectively account for over half of the phenotypes associated with these new genetic etiologies of IEI. This paper increases the number of known genetic defects identified as causing IEI to 485 (Fig. 1A, B; see all Tables and Supplementary Table 1).
Novel Inborn Errors of Immunity
Novel gene defects have been found for most categories of IEI, including novel causes of:
-
Combined immunodeficiencies (LCP2 (SLP76) [12], PAX1 [13, 14], ITPKB [15]; SASH3 [16, 17], MAN2B2 [18], COPG1 [19], IKZF2 [20,21,22,23], CHUK [24], IKZF3 [25, 26], CRACR2A [27], CD28 [28]) (Table 1; Supplementary Table 1);
-
Combined immunodeficiencies with syndromic features (MCM10 [29, 30], IL6ST [31,32,33], DIAPH1 [34]) (Table 2; Supplementary Table 1);
-
B cell deficiencies, agammaglobulinemia, or hypogammaglobulinemia (FNIP1 [35, 36], SP1I [37], PIK3CG [38, 39], POU2AF1 [40], CTNNBL1 [41], TNSRSF13 [42]) (Table 3; Supplementary Table 1);
-
Immune dysregulation (RHOG [43], SOCS1 [44,45,46], PDCD1 [47], ELF4 [48, 49], TET2 [50], CEBPE [51], IKZF1 GOF [52]) (Table 4; Supplementary Table 1)
-
innate immune defects resulting in susceptibility to mycobacterial/bacterial (TBX21 [55, 56], IFNG [57], TLR8 [58, 59]), viral (NOS2 [60], SNORA31 [61], ATG4A, MAP1LC3B2 [62], ZNFX1 [63,64,65], TLR7 [66,67,68]), and/or fungal infections (MAPK8 [69]) (Table 6; Supplementary Table 1);
-
Autoimmune/autoinflammatory disorders (TMEM173 [70], LSM11, RNU7-1 [71], CDC42 [72,73,74,75,76,77,78], STAT2 [79, 80], ATAD3A [81], AR TBK1 [82], C2orf69 [83, 84], RIPK1 [85, 86], NCKAP1L [87,88,89], SYK [90], HCK1 [91], IKBKG [92,93,94]); PSMB9 [95, 96]; and somatic variants in UBA1 [97]) (Table 7, 10, Supplementary Table 1);
-
Bone marrow failure (MECOM1) [98, 99] (Table 9; Supplementary Table 1); and
-
Phenocopies of IEI (somatic variants in TLR8 [58], autoAbs against type 1 IFNs [100,101,102,103,104]) (Table 10; Supplementary Table 1).
Novel IEI Phenocopy Known IEI, Confirming Critical Pathways for Immune Function
Some of these novel genetic findings link common clinical phenotypes that converge on a shared pathway. Examples in this update include:
-
SLP76, encoded by LCP2, is part of the TCR signalosome, interacting with or being downstream of ZAP70, LCK, LAT and ITK [105]. Thus, the phenotype of AR SLP76 deficiency overlaps substantially with that of individuals with mutations in these genes [12].
-
MCM10 is a component of the DNA replication machinery of mammalian cells and forms part of multimeric/multiprotein “replisome” complexes [106]. Thus, bi-allelic mutations in MCM10 result in a clinical phenocopy of AR MCM4 or GINS1 variants [29, 30], which also encode key proteins involved in DNA replication [106].
-
The non-redundant role of IFNγ-mediated immunity in protection against mycobacterial infection was established by identifying individuals with mutations in not only IFNG itself [57], but also TBX21 [55], the transcription factor that regulates IFNγ, who develop Mendelian susceptibility to mycobacterial disease. T-bet deficiency also resulted in upper airway inflammation and Th2 dysregulation [56], further highlighting immune regulation mediated by opposing functions of transcription factors in T cells with distinct fates (Th1 vs Th2).
-
Individuals with complete gp130-deficiency due to bi-allelic mutations of IL6ST [33], or dominant negative heterozygous variants of IL6ST [31], present with eczema, hyper-IgE, and eosinophilia, similar to individuals with AD hyper-IgE syndrome due to dominant negative mutations in STAT3 or AR mutation in ZNF341 [107]. These findings from the different genotypes indicate a key role for IL-6 signaling, via STAT3/ZNF341, in regulating hyper-IgE and atopy.
-
Store-operated calcium entry via Ca2+-release activated Ca2+ channels (CRAC) enable transfer of Ca2+ across cell membranes following activation of surface receptors, thereby eliciting Ca2+ flux and initiation of key intracellular signals [108]. Bi-allelic LOF variants in STIM1 or ORA1 disrupt Ca2+ flux, thereby impairing lymphocyte activation following engagement of antigen receptors, resulting in combined immunodeficiencies [108]. The first report of an individual with compound heterozygous inactivating variants in CRACR2A provides further insight into the importance of Ca2+-dependent signaling in immune cells [27].
-
The IKAROS family of proteins — IKAROS, AIOLOS, and HELIOS — interacts with one another as homodimers, heterodimers, or heterotrimers to regulate immune cell development and function [109]. While variants in IKZF1 encoding IKAROS have been previously reported [5, 109], individuals have now been identified with pathogenic variants in IKZF2 (HELIOS) [20,21,22,23] and IKZF3 (AIOLOS) [25, 26], as well as GOF variants in IKZF1 [52]. While these genotypes present with some distinct clinical phenotypes, there is also substantial overlap, such as B cell deficiency, hypo- or agammaglobulinemia, recurrent infections, and predisposition to B cell malignancy.
One Gene, Several Phenotypes
The discovery of novel IEI continues to demonstrate that distinct types of variants (GOF, LOF, mono-allelic, bi-allelic, exon splicing) in the same gene can cause disparate clinical conditions. This update includes AR and AD forms of IKZF2 (HELIOS) [20,21,22,23] and IL6ST [31,32,33] deficiency, as well as AD RIPK1 LOF [85, 86], AR GOF TMEM173/STING [70], AR LOF TBK1 [82], and mono-allelic IKZF1 GOF [52] variants which complement previous reports of AR RIPK1 deficiency, AD GOF TMEM173/STING, AD TBK1 deficiency, and mono-allelic IKZF1 inactivating variants, respectively [5]. AR GOF variants in CEBPE also represent a novel IEI [51]. Notably, these variants resulted in neomorphic function of the C/EBPε transcription factor, causing dysregulated expression of >400 genes, ~15–20% of which are not normally targeted by C/EBPε [51]. This may represent the prototype for neomorphic variants causing IEI.
Intriguingly, specific variants in STAT2 or IKBKG — which are already well-known to cause IEIs — have recently been reported that cause very distinct phenotypes from those previously associated with pathogenic variants in these genes. STAT2 plays a ying/yang role in type 1 IFN signalling. Thus, it is responsible for not only inducing, but also restraining, responses elicited via IFNαR1/2 complexes [110]. This regulatory role of STAT2 is mediated by binding to and recruiting USP18 to IFNαR2, which then prevents further recruitment of JAKs to type 1 IFN receptors, thereby attenuating IFNα signalling [110]. Bi-allelic variants in STAT2 that specifically affect amino acid R148 (STAT2R148Q/W) have now been reported [79, 80]. These STAT2R148Q/W variants are LOF for binding to USP18 [79, 80, 110]. Consequently, STAT2R148Q/W prevents USP18-mediated restraint of type 1 IFN signalling. It is important to appreciate that while STAT2R148Q/W is not intrinsically GOF, the net outcome of loss of STAT2-mediated regulation of type 1 IFN signalling is reminiscent of other Mendelian IFN-opathies. Indeed, STAT2R148Q/W is a phenocopy of USP18 deficiency [110], which is clearly distinct from severe susceptibility to some live attenuated viral vaccines and viral infections typical of individuals with null/nonsense mutations in STAT2 [110]. Lastly, unique variants in IKBKG that result in deletion of exon 5 were found to cause an autoinflammatory disease which is also very different from ectodermal dysplasia and immunodeficiency that is typically associated with hypomorphic IKBKG variants that impair NEMO expression and/or function [92,93,94].
Somatic/mosaic disease-causing mutations in TLR8 [58] and UBA1 [97] have also been identified, even though the pathogenic alleles were detected in only 5–30% of most blood cells (TLR8) [58] or 50–85% of myeloid cells but not in lymphocytes of fibroblasts (UBA1) [97]. These findings are an important reminder to consider the nature of genetic variants identified from unbiased next-generation sequencing, recognizing multiple mechanisms of pathogenicity for the same gene. This is highlighted by at least 40 genes having multiple entries in the current update to reflect these distinct modes of disease pathogenesis (Supplementary Table). This also emphasizes the crucial need to undertake in-depth in vitro functional validation of any variant considered to be potentially pathogenic. Alternatively, it signifies the difficulty in excluding a candidate pathogenic variant without functional testing. It also underscores the need to consider variants detected at low allelic frequencies that may represent somatic/mosaic, rather than germline, variants. These findings also predict that somatic variants in key immune genes will be frequently discovered as causes of novel IEI in the not-too distant future [111].
IEI Define Specific Roles for Known Genes and Reveal Immune-Specific Functions of Novel Genes
One of most profound outcomes of discovering the genetic cause of an IEI is the ability to ascribe unequivocally non-redundant, as well as redundant, functions to a specific gene in human immunity. Classic examples of this are the fundamental requirement for IL2RG in humans for the development of T and NK cells, but not B cells, and the essential role of STAT3 for CD4+ T cell differentiation into Th17 cells and subsequent host defense against fungal infections, but not for the generation of most other CD4+ T cell effector populations [112]. Findings included in this update confirm data from mice on the importance of FNIP1 and SPI1 (encoding PU.1) during human B cell development [35,36,37] and the fundamental regulatory role of PD-1 (encoded by PDCD1) in human immune function [47]. However, and perhaps counter to all expectations and immunology dogma relating to T cell co-stimulation, CD28 is required for host defense against HPV but is largely redundant in the face of other infectious pathogens [28]. Who would have thought!
The latest IEI have also revealed critical roles for genes not previously strongly associated with immune regulation and/or host defense. For instance, we have now learned that:
-
The SH3-domain containing protein SASH3 contributes to B and T cell developments [16, 17].
-
ZNFX1, a member of an RNA helicase superfamily, plays a dual role in human immunity, including in innate immune responses against viruses, bacteria, mycobacteria, and fungi, as well as in restraining type 1 IFN-mediated inflammation [63,64,65].
-
The small nucleolar RNA SNORA31 plays a critical role in CNS-intrinsic immunity against HSV-2 infection, likely via production of type 1 IFN, yet the exact mechanism remains unknown [61].
-
The hitherto uncharacterized protein-coding gene C2orf69 has a multitude of roles across numerous biological systems, including regulating autoinflammation [83, 84].
The discovery of these novel IEIs provides opportunities to further extend our understanding of human immunity and immune regulation.
SARS-CoV2 and Inborn Errors of Immunity
The emergence of novel pathogens poses potential health risks to the general population due to the lack of substantial pre-existing immune memory. More critically though, individuals with specific germline genetic variants — causing known and unknown IEIs — may be at greater risk of experiencing more severe disease following infection than the general population. The COVID-19 pandemic has indeed revealed genes and pathways essential for anti-SARS-CoV2 immunity. Genomic studies discovered that ~2–3% of cases of severe life-threatening SARS-CoV2 infection resulted from germline LOF/LOE variants in the type 1 IFN signaling pathway: TLR3, UNC93B1, TICAM1, TBK1, IRF3, IRF7, IFNAR1, and IFNAR2 [113]. These findings are reminiscent of earlier studies that identified variants in these genes in individuals susceptible to life-threatening infections with other viruses, including influenza virus, HSV-1, and live viral vaccines [114]. Hemizygous deleterious variants have also been identified in TLR7 in ~1% of males who developed severe/fatal COVID-19 [66,67,68]. Thus, X-linked TLR7 deficiency represents a novel IEI predisposing to severe COVID-19.
The importance of type 1 IFN in anti-SARS-CoV2 immunity was also realized by the finding that ~10–20% of patients with severe COVID-19 have high levels of neutralizing serum autoantibodies (autoAbs) against type 1 IFNs; these were not detected in asymptomatic infected individuals [100,101,102,103,104]. Collectively, these studies defined a non-redundant role for type 1 IFNs in host defense against SARS-CoV2 infection and established that autoAbs against type 1 IFN phenocopy an IEI.
Conclusions
The goals of the IUIS Expert Committee on IEI are to increase awareness, facilitate recognition, promote optimal treatment, and support research in the field of clinical immunology. Since the last IEI update, we have continued to witness the ongoing rapid identification, and molecular, biochemical, and cellular characterization, of genetic variants that cause human diseases by disrupting host defense or immune regulation. The 55 novel gene defects reported here bring to total number of IEI to 485 (Fig. 1A, B), thus underscoring the power of next-generation sequencing technologies and sophisticated functional validation of candidate pathogenic variants to (1) identify novel gene defects underlying human disease, (2) elucidate mechanisms of disease pathogenesis, (3) define non-redundant functions of key genes in human immune cell development, host defense and immune regulation, (4) expand the immunological and clinical phenotypes of IEI, and (5) implement gene-specific therapies. These fundamental discoveries continue to highlight the critical contributions of IEI to our broader understanding of basic, translational, and clinical immunology, as well as molecular medicine. And we will no doubt observe novel insights into basic and clinical immunology with the next wave of novel IEIs.
Data Availability
Not applicable
References
Zhang Q, Frange P, Blanche S, Casanova JL. Pathogenesis of infections in HIV-infected individuals: insights from primary immunodeficiencies. Curr Opin Immunol. 2017;48:122–33. https://doi.org/10.1016/j.coi.2017.09.002.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128. https://doi.org/10.1007/s10875-017-0464-9.
Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2018;38(1):129–43. https://doi.org/10.1007/s10875-017-0465-8.
Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS Phenotypical Classification. J Clin Immunol. 2020;40(1):66–81. https://doi.org/10.1007/s10875-020-00758-x.
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. https://doi.org/10.1007/s10875-019-00737-x.
Casanova JL, Abel L. Human genetics of infectious diseases: unique insights into immunological redundancy. Semin Immunol. 2018;36:1–12. https://doi.org/10.1016/j.smim.2017.12.008.
Fischer A, Rausell A. What do primary immunodeficiencies tell us about the essentiality/redundancy of immune responses? Semin Immunol. 2018;36:13–6. https://doi.org/10.1016/j.smim.2017.12.001.
Picard C, Fischer A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol. 2014;44(10):2854–61. https://doi.org/10.1002/eji.201444669.
Leiding JW, Forbes LR. Mechanism-based precision therapy for the treatment of primary immunodeficiency and primary immunodysregulatory diseases. J Allergy Clin Immunol Pract. 2019;7(3):761–73. https://doi.org/10.1016/j.jaip.2018.12.017.
Ma CS, Tangye SG. Flow cytometric-based analysis of defects in lymphocyte differentiation and function due to inborn errors of immunity. Front Immunol. 2019;10:2108. https://doi.org/10.3389/fimmu.2019.02108.
Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211(11):2137–49. https://doi.org/10.1084/jem.20140520.
Lev A, Lee YN, Sun G, Hallumi E, Simon AJ, Zrihen KS, et al. Inherited SLP76 deficiency in humans causes severe combined immunodeficiency, neutrophil and platelet defects. J Exp Med. 2021;218(3). https://doi.org/10.1084/jem.20201062.
Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. 2020;5(44). https://doi.org/10.1126/sciimmunol.aax1036.
Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. 2017;92(6):664–8. https://doi.org/10.1111/cge.13085.
Almutairi A, Wallace JG, Jaber F, Alosaimi MF, Jones J, Sallam MTH, et al. Severe combined immunodeficiency caused by inositol-trisphosphate 3-kinase B (ITPKB) deficiency. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.01.014.
Delmonte OM, Bergerson JRE, Kawai T, Kuehn HS, McDermott DH, Cortese I, et al. SASH3 variants cause a novel form of X-linked combined immunodeficiency with immune dysregulation. Blood. 2021;138(12):1019–33. https://doi.org/10.1182/blood.2020008629.
Labrador-Horrillo M, Franco-Jarava C, Garcia-Prat M, Parra-Martinez A, Antolin M, Salgado-Perandres S, et al. Case report: X-Linked SASH3 deficiency presenting as a common variable immunodeficiency. Front Immunol. 2022;13:881206. https://doi.org/10.3389/fimmu.2022.881206.
Verheijen J, Wong SY, Rowe JH, Raymond K, Stoddard J, Delmonte OM, et al. Defining a new immune deficiency syndrome: MAN2B2-CDG. J Allergy Clin Immunol. 2020;145(3):1008–11. https://doi.org/10.1016/j.jaci.2019.11.016.
Bainter W, Platt CD, Park SY, Stafstrom K, Wallace JG, Peters ZT, et al. Combined immunodeficiency due to a mutation in the gamma1 subunit of the coat protein I complex. J Clin Invest. 2021;131(3). https://doi.org/10.1172/JCI140494.
Hetemaki I, Kaustio M, Kinnunen M, Heikkila N, Keskitalo S, Nowlan K, et al. Loss-of-function mutation in IKZF2 leads to immunodeficiency with dysregulated germinal center reactions and reduction of MAIT cells. Sci Immunol. 2021;6(65):eabe3454. https://doi.org/10.1126/sciimmunol.abe3454.
Shahin T, Kuehn HS, Shoeb MR, Gawriyski L, Giuliani S, Repiscak P, et al. Germline biallelic mutation affecting the transcription factor Helios causes pleiotropic defects of immunity. Sci Immunol. 2021;6(65):eabe3981. https://doi.org/10.1126/sciimmunol.abe3981.
Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magerus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. 2019;134(1):9–21. https://doi.org/10.1182/blood-2018-11-887141.
Shahin T, Mayr D, Shoeb MR, Kuehn HS, Hoeger B, Giuliani S, et al. Identification of germline monoallelic mutations in IKZF2 in patients with immune dysregulation. Blood Adv. 2021. https://doi.org/10.1182/bloodadvances.2021006367.
Bainter W, Lougaris V, Wallace JG, Badran Y, Hoyos-Bachiloglu R, Peters Z, et al. Combined immunodeficiency with autoimmunity caused by a homozygous missense mutation in inhibitor of nuclear factor B kinase alpha (IKKalpha). Sci Immunol. 2021;6(63):eabf6723. https://doi.org/10.1126/sciimmunol.abf6723.
Yamashita M, Kuehn HS, Okuyama K, Okada S, Inoue Y, Mitsuiki N, et al. A variant in human AIOLOS impairs adaptive immunity by interfering with IKAROS. Nat Immunol. 2021;22(7):893–903. https://doi.org/10.1038/s41590-021-00951-z.
Kuehn HS, Chang J, Yamashita M, Niemela JE, Zou C, Okuyama K, et al. T and B cell abnormalities, pneumocystis pneumonia, and chronic lymphocytic leukemia associated with an AIOLOS defect in patients. J Exp Med. 2021;218(12). https://doi.org/10.1084/jem.20211118.
Wu B, Rice L, Shrimpton J, Lawless D, Walker K, Carter C, et al. Biallelic mutations in calcium release activated channel regulator 2A (CRACR2A) cause a primary immunodeficiency disorder. Elife. 2021;10. https://doi.org/10.7554/eLife.72559.
Beziat V, Rapaport F, Hu J, Titeux M, Bonnet des Claustres M, Bourgey M et al. Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell. 2021;184(14):3812-28 e30. doi:https://doi.org/10.1016/j.cell.2021.06.004.
Mace EM, Paust S, Conte MI, Baxley RM, Schmit MM, Patil SL, et al. Human NK cell deficiency as a result of biallelic mutations in MCM10. J Clin Invest. 2020. https://doi.org/10.1172/JCI134966.
Baxley RM, Leung W, Schmit MM, Matson JP, Yin L, Oram MK, et al. Bi-allelic MCM10 variants associated with immune dysfunction and cardiomyopathy cause telomere shortening. Nat Commun. 2021;12(1):1626. https://doi.org/10.1038/s41467-021-21878-x.
Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. 2020;217(6). https://doi.org/10.1084/jem.20191804.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104(6):1182–201. https://doi.org/10.1016/j.ajhg.2019.04.011.
Chen YH, Grigelioniene G, Newton PT, Gullander J, Elfving M, Hammarsjo A, et al. Absence of GP130 cytokine receptor signaling causes extended Stuve-Wiedemann syndrome. J Exp Med. 2020;217(3). https://doi.org/10.1084/jem.20191306.
Kaustio M, Nayebzadeh N, Hinttala R, Tapiainen T, Astrom P, Mamia K, et al. Loss of DIAPH1 causes SCBMS, combined immunodeficiency, and mitochondrial dysfunction. J Allergy Clin Immunol. 2021;148(2):599–611. https://doi.org/10.1016/j.jaci.2020.12.656.
Niehues T, Ozgur TT, Bickes M, Waldmann R, Schoning J, Brasen J, et al. Mutations of the gene FNIP1 associated with a syndromic autosomal recessive immunodeficiency with cardiomyopathy and pre-excitation syndrome. Eur J Immunol. 2020;50(7):1078–80. https://doi.org/10.1002/eji.201948504.
Saettini F, Poli C, Vengoechea J, Bonanomi S, Orellana JC, Fazio G, et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin interacting protein 1 deficiency. Blood. 2020. https://doi.org/10.1182/blood.2020006441.
Le Coz C, Nguyen DN, Su C, Nolan BE, Albrecht AV, Xhani S, et al. Constrained chromatin accessibility in PU.1-mutated agammaglobulinemia patients. J Exp Med. 2021;218(7). https://doi.org/10.1084/jem.20201750.
Takeda AJ, Maher TJ, Zhang Y, Lanahan SM, Bucklin ML, Compton SR, et al. Human PI3Kgamma deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat Commun. 2019;10(1):4364. https://doi.org/10.1038/s41467-019-12311-5.
Thian M, Hoeger B, Kamnev A, Poyer F, Kostel Bal S, Caldera M, et al. Germline biallelic PIK3CG mutations in a multifaceted immunodeficiency with immune dysregulation. Haematologica. 2020. https://doi.org/10.3324/haematol.2019.231399.
Kury P, Staniek J, Wegehaupt O, Janowska I, Eckenweiler M, Korinthenberg R, et al. Agammaglobulinemia with normal B-cell numbers in a patient lacking Bob1. J Allergy Clin Immunol. 2021;147(5):1977–80. https://doi.org/10.1016/j.jaci.2021.01.027.
Kuhny M, Forbes LR, Cakan E, Vega-Loza A, Kostiuk V, Dinesh RK, et al. Disease-associated CTNNBL1 mutation impairs somatic hypermutation by decreasing nuclear AID. J Clin Invest. 2020. https://doi.org/10.1172/JCI131297.
Yeh TW, Okano T, Naruto T, Yamashita M, Okamura M, Tanita K, et al. APRIL-dependent life-long plasmacyte maintenance and immunoglobulin production in humans. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.03.025.
Kalinichenko A, Perinetti Casoni G, Dupre L, Trotta L, Huemer J, Galgano D, et al. RhoG deficiency abrogates cytotoxicity of human lymphocytes and causes hemophagocytic lymphohistiocytosis. Blood. 2021;137(15):2033–45. https://doi.org/10.1182/blood.2020008738.
Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.07.033.
Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583(7814):90–5. https://doi.org/10.1038/s41586-020-2265-1.
Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. 2020;11(1):5341. https://doi.org/10.1038/s41467-020-18925-4.
Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat Med. 2021;27(9):1646–54. https://doi.org/10.1038/s41591-021-01388-5.
Tyler PM, Bucklin ML, Zhao M, Maher TJ, Rice AJ, Ji W, et al. Human autoinflammatory disease reveals ELF4 as a transcriptional regulator of inflammation. Nat Immunol. 2021;22(9):1118–26. https://doi.org/10.1038/s41590-021-00984-4.
Sun G, Qiu L, Yu L, An Y, Ding Y, Zhou L, et al. Loss of function mutation in ELF4 causes autoinflammatory and immunodeficiency disease in human. J Clin Immunol. 2022. https://doi.org/10.1007/s10875-022-01243-3.
Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood. 2020;136(9):1055–66. https://doi.org/10.1182/blood.2020005844.
Goos H, Fogarty CL, Sahu B, Plagnol V, Rajamaki K, Nurmi K, et al. Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J Allergy Clin Immunol. 2019;144(5):1364–76. https://doi.org/10.1016/j.jaci.2019.06.003.
Hoshino A, Boutboul D, Zhang Y, Kuehn HS, Hadjadj J, Ozdemir N, et al. Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci Immunol. 2022;7(69):eabi7160. https://doi.org/10.1126/sciimmunol.abi7160.
Marin-Esteban V, Youn J, Beaupain B, Jaracz-Ros A, Barlogis V, Fenneteau O, et al. Biallelic CXCR2 loss-of-function mutations define a distinct congenital neutropenia entity. Haematologica. 2021. https://doi.org/10.3324/haematol.2021.279254.
Auer PL, Teumer A, Schick U, O’Shaughnessy A, Lo KS, Chami N, et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet. 2014;46(6):629–34. https://doi.org/10.1038/ng.2962.
Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, et al. Human T-bet governs innate and innate-like adaptive IFN-gamma immunity against mycobacteria. Cell. 2020;183(7):1826–47 e31. https://doi.org/10.1016/j.cell.2020.10.046.
Yang R, Weisshaar M, Mele F, Benhsaien I, Dorgham K, Han J, et al. High Th2 cytokine levels and upper airway inflammation in human inherited T-bet deficiency. J Exp Med. 2021;218(8). https://doi.org/10.1084/jem.20202726.
Kerner G, Rosain J, Guerin A, AlKhabaz A, Oleaga-Quintas C, Rapaport F, et al. Inherited human IFNgamma deficiency underlies mycobacterial disease. J Clin Invest. 2020. https://doi.org/10.1172/JCI135460.
Aluri J, Bach A, Kaviany S, Chiquetto Paracatu L, Kitcharoensakkul M, Walkiewicz MA, et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood. 2021;137(18):2450–62. https://doi.org/10.1182/blood.2020009620.
Fejtkova M, Sukova M, Hlozkova K, Skvarova Kramarzova K, Rackova M, Jakubec D, et al. TLR8/TLR7 dysregulation due to a novel TLR8 mutation causes severe autoimmune hemolytic anemia and autoinflammation in identical twins. Am J Hematol. 2022;97(3):338–51. https://doi.org/10.1002/ajh.26452.
Drutman SB, Mansouri D, Mahdaviani SA, Neehus AL, Hum D, Bryk R, et al. Fatal cytomegalovirus infection in an adult with inherited NOS2 deficiency. N Engl J Med. 2020;382(5):437–45. https://doi.org/10.1056/NEJMoa1910640.
Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, et al. Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat Med. 2019;25(12):1873–84. https://doi.org/10.1038/s41591-019-0672-3.
Hait AS, Olagnier D, Sancho-Shimizu V, Skipper KA, Helleberg M, Larsen SM, et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci Immunol. 2020;5(54). https://doi.org/10.1126/sciimmunol.abc2691.
Vavassori S, Chou J, Faletti LE, Haunerdinger V, Opitz L, Joset P, et al. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J Allergy Clin Immunol. 2021;148(2):381–93. https://doi.org/10.1016/j.jaci.2021.03.045.
Le Voyer T, Neehus AL, Yang R, Ogishi M, Rosain J, Alroqi F, et al. Inherited deficiency of stress granule ZNFX1 in patients with monocytosis and mycobacterial disease. Proc Natl Acad Sci U S A. 2021;118(15). https://doi.org/10.1073/pnas.2102804118.
Alawbathani S, Westenberger A, Ordonez-Herrera N, Al-Hilali M, Al Hebby H, Alabbas F, et al. Biallelic ZNFX1 variants are associated with a spectrum of immuno-hematological abnormalities. Clin Genet. 2022;101(2):247–54. https://doi.org/10.1111/cge.14081.
Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, Maglorius Renkilaraj MRL, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6(62). https://doi.org/10.1126/sciimmunol.abl4348.
van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324(7):663–73. https://doi.org/10.1001/jama.2020.13719.
Abolhassani H, Vosughimotlagh A, Asano T, Landegren N, Boisson B, Delavari S, et al. X-linked TLR7 deficiency underlies critical COVID-19 pneumonia in a male patient with ataxia-telangiectasia. J Clin Immunol. 2021. https://doi.org/10.1007/s10875-021-01151-y.
Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol. 2019;4(41). https://doi.org/10.1126/sciimmunol.aax7965.
Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI). J Allergy Clin Immunol. 2020;146(5):1204–8 e6. https://doi.org/10.1016/j.jaci.2020.06.032.
Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. 2020;52(12):1364–72. https://doi.org/10.1038/s41588-020-00737-3.
Verboon JM, Mahmut D, Kim AR, Nakamura M, Abdulhay NJ, Nandakumar SK, et al. Infantile myelofibrosis and myeloproliferation with CDC42 dysfunction. J Clin Immunol. 2020. https://doi.org/10.1007/s10875-020-00778-7.
Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. 2019;216(12):2778–99. https://doi.org/10.1084/jem.20190147.
Gernez Y, de Jesus AA, Alsaleem H, Macaubas C, Roy A, Lovell D, et al. Severe autoinflammation in 4 patients with C-terminal variants in cell division control protein 42 homolog (CDC42) successfully treated with IL-1beta inhibition. J Allergy Clin Immunol. 2019;144(4):1122–5 e6. https://doi.org/10.1016/j.jaci.2019.06.017.
Bucciol G, Pillay B, Casas-Martin J, Delafontaine S, Proesmans M, Lorent N, et al. Systemic inflammation and myelofibrosis in a patient with Takenouchi-Kosaki syndrome due to CDC42 Tyr64Cys mutation. J Clin Immunol. 2020. https://doi.org/10.1007/s10875-020-00742-5.
Bekhouche B, Tourville A, Ravichandran Y, Tacine R, Abrami L, Dussiot M, et al. A toxic palmitoylation of Cdc42 enhances NF-kappaB signaling and drives a severe autoinflammatory syndrome. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.03.020.
He T, Huang Y, Ling J, Yang J. A new patient with NOCARH syndrome due to CDC42 defect. J Clin Immunol. 2020;40(4):571–5. https://doi.org/10.1007/s10875-020-00786-7.
Szczawinska-Poplonyk A, Ploski R, Bernatowska E, Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: a case report. Front Immunol. 2020;11:318. https://doi.org/10.3389/fimmu.2020.00318.
Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol 2019;4(42). https://doi.org/10.1126/sciimmunol.aav7501.
Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med. 2020;217(5). https://doi.org/10.1084/jem.20192319.
Lepelley A, Della Mina E, Van Nieuwenhove E, Waumans L, Fraitag S, Rice GI, et al. Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A. J Exp Med. 2021;218(10). https://doi.org/10.1084/jem.20201560.
Taft J, Markson M, Legarda D, Patel R, Chan M, Malle L, et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell. 2021;184(17):4447–63 e20. https://doi.org/10.1016/j.cell.2021.07.026.
Wong HH, Seet SH, Maier M, Gurel A, Traspas RM, Lee C, et al. Loss of C2orf69 defines a fatal autoinflammatory syndrome in humans and zebrafish that evokes a glycogen-storage-associated mitochondriopathy. Am J Hum Genet. 2021;108(7):1301–17. https://doi.org/10.1016/j.ajhg.2021.05.003.
Lausberg E, Giesselmann S, Dewulf JP, Wiame E, Holz A, Salvarinova R, et al. C2orf69 mutations disrupt mitochondrial function and cause a multisystem human disorder with recurring autoinflammation. J Clin Invest. 2021;131(12). https://doi.org/10.1172/JCI143078.
Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577(7788):109–14. https://doi.org/10.1038/s41586-019-1830-y.
Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577(7788):103–8. https://doi.org/10.1038/s41586-019-1828-5.
Cook SA, Comrie WA, Poli MC, Similuk M, Oler AJ, Faruqi AJ, et al. HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease. Science. 2020;369(6500):202–7. https://doi.org/10.1126/science.aay5663.
Salzer E, Zoghi S, Kiss MG, Kage F, Rashkova C, Stahnke S, et al. The cytoskeletal regulator HEM1 governs B cell development and prevents autoimmunity. Sci Immunol. 2020;5(49). https://doi.org/10.1126/sciimmunol.abc3979.
Castro CN, Rosenzwajg M, Carapito R, Shahrooei M, Konantz M, Khan A, et al. NCKAP1L defects lead to a novel syndrome combining immunodeficiency, lymphoproliferation, and hyperinflammation. J Exp Med. 2020;217(12). https://doi.org/10.1084/jem.20192275.
Wang L, Aschenbrenner D, Zeng Z, Cao X, Mayr D, Mehta M, et al. Gain-of-function variants in SYK cause immune dysregulation and systemic inflammation in humans and mice. Nat Genet. 2021;53(4):500–10. https://doi.org/10.1038/s41588-021-00803-4.
Kanderova V, Svobodova T, Borna S, Fejtkova M, Martinu V, Paderova J, et al. Early-onset pulmonary and cutaneous vasculitis driven by constitutively active SRC-family kinase HCK. J Allergy Clin Immunol. 2021. https://doi.org/10.1016/j.jaci.2021.07.046.
de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. 2020;130(4):1669–82. https://doi.org/10.1172/JCI129301.
Hegazy S, Marques MC, Canna SW, Goldbach-Mansky R, de Jesus AA, Reyes-Mugica M, et al. NEMO-NDAS: a panniculitis in the young representing an autoinflammatory disorder in disguise. Am J Dermatopathol. 2022. https://doi.org/10.1097/DAD.0000000000002144.
Lee Y, Wessel AW, Xu J, Reinke JG, Lee E, Kim SM, et al. Genetically programmed alternative splicing of NEMO mediates an autoinflammatory disease phenotype. J Clin Invest. 2022;132(6). https://doi.org/10.1172/JCI128808.
Kataoka S, Kawashima N, Okuno Y, Muramatsu H, Miwata S, Narita K, et al. Successful treatment of a novel type I interferonopathy due to a de novo PSMB9 gene mutation with a Janus kinase inhibitor. J Allergy Clin Immunol. 2021;148(2):639–44. https://doi.org/10.1016/j.jaci.2021.03.010.
Kanazawa N, Hemmi H, Kinjo N, Ohnishi H, Hamazaki J, Mishima H, et al. Heterozygous missense variant of the proteasome subunit beta-type 9 causes neonatal-onset autoinflammation and immunodeficiency. Nat Commun. 2021;12(1):6819. https://doi.org/10.1038/s41467-021-27085-y.
Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2026834.
Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet. 2015;97(6):848–54. https://doi.org/10.1016/j.ajhg.2015.10.010.
Germeshausen M, Ancliff P, Estrada J, Metzler M, Ponstingl E, Rutschle H, et al. MECOM-associated syndrome: a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018;2(6):586–96. https://doi.org/10.1182/bloodadvances.2018016501.
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515). https://doi.org/10.1126/science.abd4585.
Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol. 2021;6(62). https://doi.org/10.1126/sciimmunol.abl4340.
Abers MS, Rosen LB, Delmonte OM, Shaw E, Bastard P, Imberti L, et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol Cell Biol. 2021;99(9):917–21. https://doi.org/10.1111/imcb.12495.
Troya J, Bastard P, Planas-Serra L, Ryan P, Ruiz M, de Carranza M, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in Madrid, Spain. J Clin Immunol. 2021;41(5):914–22. https://doi.org/10.1007/s10875-021-01036-0.
Solanich X, Rigo-Bonnin R, Gumucio VD, Bastard P, Rosain J, Philippot Q, et al. Pre-existing autoantibodies neutralizing high concentrations of type I interferons in almost 10% of COVID-19 patients admitted to intensive care in Barcelona. J Clin Immunol. 2021;41(8):1733–44. https://doi.org/10.1007/s10875-021-01136-x.
Koretzky GA, Abtahian F, Silverman MA. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat Rev Immunol. 2006;6(1):67–78. https://doi.org/10.1038/nri1750.
Bellelli R, Boulton SJ. Spotlight on the replisome: aetiology of dna replication-associated genetic diseases. Trends Genet. 2021;37(4):317–36. https://doi.org/10.1016/j.tig.2020.09.008.
Chen YH, Spencer S, Laurence A, Thaventhiran JE, Uhlig HH. Inborn errors of IL-6 family cytokine responses. Curr Opin Immunol. 2021;72:135–45. https://doi.org/10.1016/j.coi.2021.04.007.
Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79. https://doi.org/10.1111/nyas.12938.
Yamashita M, Morio T. Inborn errors of IKAROS and AIOLOS. Curr Opin Immunol. 2021;72:239–48. https://doi.org/10.1016/j.coi.2021.06.010.
Duncan CJA, Hambleton S. Human disease phenotypes associated with loss and gain of function mutations in STAT2: viral susceptibility and type I interferonopathy. J Clin Immunol. 2021;41(7):1446–56. https://doi.org/10.1007/s10875-021-01118-z.
Van Horebeek L, Dubois B, Goris A. Somatic variants: new kids on the block in human immunogenetics. Trends Genet. 2019;35(12):935–47. https://doi.org/10.1016/j.tig.2019.09.005.
Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–28. https://doi.org/10.1016/j.immuni.2012.03.016.
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515). https://doi.org/10.1126/science.abd4570.
Moens L, Meyts I. Recent human genetic errors of innate immunity leading to increased susceptibility to infection. Curr Opin Immunol. 2020;62:79–90. https://doi.org/10.1016/j.coi.2019.12.002.
Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015;125(11):4196–211. https://doi.org/10.1172/JCI81260.
Funding
The Inborn Errors of Immunity Expert Committee received funding from the International Union of Immunological Societies; CSL Behring, Baxalta, and Shire/Takeda provided educational grants to enable us to compile this interim update to novel causes of immune diseases. This work was also supported in part by the Intramural Research Program of the NIAID, NIH. SGT is supported by an Investigator Grant (Level 3) awarded by the National Health and Medical Research Council of Australia. IM is a senior clinical investigator of FWO Vlaanderen (EBD-D8974-FKM)
Author information
Authors and Affiliations
Contributions
SGT wrote the drafts of the manuscript, prepared the tables, and revised the original manuscripts for resubmission. All co-authors contributed to and edited drafts of the original and revised manuscripts and tables and approved the final submitted version.
Corresponding author
Ethics declarations
Ethics Approval
This work is a summary of recently reported genetic variants that represent novel inborn errors of immunity. No human research studies were performed to produce this summary. Thus, no approvals by appropriate institutional review boards or human research ethics committees were required to undertake the preparation of this report.
Consent to Participate
Not applicable.
Consent for Publication
The authors consent to publish the content of this summary. However, as noted above, as this is a summary of recently-reported genetic variants that represent novel inborn errors of immunity, we did not require consent to publish from participants.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tangye, S.G., Al-Herz, W., Bousfiha, A. et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 42, 1473–1507 (2022). https://doi.org/10.1007/s10875-022-01289-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01289-3