
Abstract
Background
Inborn errors of immunity (IEI) are a group of heterogeneous disorders with geographic and ethnic diversities. Although IEI are common in Egypt, genetic diagnosis is limited due to financial restrictions. This study aims to characterize the genetic spectrum of IEI patients in Egypt and highlights the adaptation of the molecular diagnostic methods to a resource-limited setting.
Methods
Genetic material from 504 patients was studied, and proper diagnosis was achieved in 282 patients from 246 families. Mutational analysis was done by Sanger sequencing, next-generation sequencing (NGS) targeting customized genes panels, and whole-exome sequencing (WES) according to the patients’ phenotypes and availability of genetic testing.
Results
A total of 194 variants involving 72 different genes were detected with RAG1/2 genes being the most encountered followed by DOCK8, CYBA, LRBA, NCF1, and JAK3. Autosomal recessive (AR) inheritance was detected in 233/282 patients (82.6%), X-linked (XL) recessive inheritance in 32/282 patients (11.3%), and autosomal dominant (AD) inheritance in 18/282 patients (6.4%), reflecting the impact of consanguineous marriages on the prevalence of different modes of inheritance and the distribution of the various IEI disorders.
Conclusion
The study showed that a combination of Sanger sequencing in selected patients associated with targeted NGS or WES in other patients is an effective diagnostic strategy for IEI diagnosis in countries with limited diagnostic resources. Molecular testing can be used to validate other nonexpensive laboratory techniques that help to reach definitive diagnosis and help in genetic counseling and taking proper therapeutic decisions including stem cell transplantation or gene therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inborn errors of immunity (IEI) comprise a heterogeneous group of genetic disorders caused by defects in one or more components of the immune system, resulting in a wide spectrum of clinical manifestations and laboratory abnormalities. These patients have an increased susceptibility to infections and higher risks of developing autoimmune diseases and malignancies [1, 2].
A delay in the diagnosis can contribute to the morbidity and mortality, especially the atypical cases where genetic testing becomes an indispensable part of patients’ evaluation. It provides a definitive diagnosis, helps to establish phenotype-genotype correlation, improves decisions of curative interventions, and opens the possibility of genetic counseling for affected families [2, 3].
This study aims to outline the genetic makeup of IEI diseases in Egypt (a country with high rates of consanguinity and limited resources) and describes the achievement in the field of genetic diagnosis of IEI in Egypt in the past decade and the optimum way to use the available genetic tools to reach a refined precise diagnosis.
Patients and Methods
Among 1496 IEI patients referred between 2010 and 2021 to the Tertiary Primary Immunodeficiency (PID) Referral Center at Cairo University specialized Children’s Hospital (Cairo, Egypt), 1000 patients with specific phenotype/probable diagnosis of IEI according to International Union of Immunological Societies (IUIS) classification [4]/ESID criteria for diagnosis [5] were included for genetic analysis in this study. An informed consent was taken from the patients’ parents or legal guardians and the study was approved by the local institutional review board.
Detailed medical history, clinical evaluation, and laboratory workup were recorded for each patient. Genomic DNA was extracted from the peripheral blood using QIAamp DNA blood Minikit (Qiagen, Germany) according to the manufacturer’s instructions.
Sanger sequencing using earlier established protocols was performed for patients with classical clinical presentations denoting a specific PID phenotype; e.g., severe combined immunodeficiency disorder (SCID) were tested for RAG1, RAG2, PNP, ADA, JAK3, IL7RG genes based on flow cytometry results; chronic granulomatous disease (CGD) were tested for CYBA, NCF1, NCF2 genes guided by the results of flow cytometry for the defective intracellular proteins; very early onset inflammatory bowel disease (VEO-IBD) tested for IL10RA and IL10RB, and leucocyte adhesion deficiency (LAD) patients with defective CD18 expression were tested for ITGB2 gene.
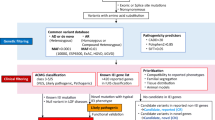
Meanwhile, for patients with atypical phenotypes and for those with distinct phenotype, however, Sanger sequencing is not applicable (e.g., large genes), or in case of absence of pathogenic variant in the targeted candidate gene tested by Sanger; NGS or WES was done based on the circumstantial test availability (Fig. 1). For Sanger sequencing, amplification was carried out by polymerase chain reaction (PCR) using designed primers targeting all the exons and exon–intron junctions. PCR products were sequenced on Applied Biosystems™ 3500 Genetic Analyzer (Applied Biosystems, USA) utilizing the same primers used for PCR fragment amplification. Sequences were compared with the reference sequence published by the National Centre for Biotechnology Information and analyzed using the Basic Local Alignment Search Tool (BLAST).

The followed algorithm for genetic testing of IEI according to patients’ phenotypes
Meanwhile, for next-generation sequencing (NGS), the DNA was used to perform targeted gene capture using a custom capture kit. The panel was designed to cover the exons, exon–intron junctions, and UTRs.
For whole-exome sequencing (WES), Agilent’s SureSelect All Exon V6 + UTR kit was used on a HiSeq4000 system (Illumina, San Diego, CA). The raw reads were first cleaned by removing adapter sequences, trimming low-quality ends, and filtering reads with low quality (Phred quality < 20). The high-quality reads were aligned with the human genome (GRCh37) using Bowtie2 (version 2.3.2, Johns Hopkins University, Baltimore).
Prediction of functional effects of amino acid substitutions was performed using the Polymorphism Phenotyping version 2 software tool (PolyPhen-2) and sorting intolerant from tolerant (SIFT) software. The clinical significance of reported variants and the genotype–phenotype correlation were assessed by protein variation effect analyzer (PROVEAN), VARSOME, and clinically relevant variant (ClinVar) database. Accordingly, the variants were labeled as pathogenic/likely pathogenic/variant of uncertain significance (VUS)/likely benign/benign.
Results
Among 1000 patients with specific phenotype/probable diagnosis of IEI, a group of 483 Familial Mediterranean fever (FMF) patients and 13 cystic fibrosis patients were diagnosed by strip hybridization assay method and excluded from the study as these patients were not followed up at the PID clinic. Results of only 504 IEI patients are discussed hereby (Supplemental Table 1).
Genetic diagnosis was reached in 282/504 patients (56%) from 246 families: 200 consanguineous (81.3%) and 46 non-consanguineous families (18.7%). We could not detect pathogenic variants that can explain the clinical phenotype in the remaining 222 patients (44%). The diagnosed patients were 171 males (60.6%) including 30 patients with XL-IEI and 111 females (39.4%).
Variants were identified by Sanger sequencing in 108/282 patients (38.3%), NGS in 102/282 patients (36.2%), and WES in 68/282 patients (24.1%). Fluorescent in situ hybridization (FISH) was used for the diagnosis of 4 patients (1.4%) with DiGeorge syndrome. A total of 231 patients were tested to begin with by Sanger sequencing with a diagnostic yield of 46.8% (108/231), 282 were tested by NGS/WES with a diagnostic yield of 60.3% (170/282), including 13 patients who were tested by NGS/WES following the failure of Sanger sequencing to identify a pathogenic variant in tested gene (Fig. 2).

An algorithm representing the total number of IEI patients followed up in Cairo University Children Hospital, the percentage of patients subjected for genetic diagnosis, and the different molecular techniques used to reach definitive diagnosis
Mutations were identified in 72 different genes; RAG1/2, DOCK8, CYBA, LRBA, NCF1, and JAK3 genes were the most encountered being detected in 43, 24, 22, 21, 13, and 12 patients respectively (Fig. 3).

The genetic makeup of IEI patients and the frequency of encountering each gene
Two hundred thirty-three patients, 233/282 (82.6%), inherited the mutations in an autosomal recessive (AR) pattern with 6 patients having two affected genes simultaneously, 32/282 patients (11.3%) in an X-linked (XL) pattern while 18/282 patients (6.4%) had an autosomal dominant (AD) disease. Six patients had hits in two different IEI genes; this included the following: three patients with disease causing variants in both DOCK8 gene and CARD9 gene, 1 patient with pathogenic variant in DOCK8 gene associated with compound heterozygous variants in AIRE gene, 2 patients with pathogenic variants in LRBA gene and associated variant in MSH6 gene. One patient had pathogenic variants in two genes with different modes of inheritance: HUWE1 gene (XL) and PLCG2 gene (AD). Another six unrelated patients were found to carry the same two simultaneous homozygous variants in RAG2 gene p.Thr215Ile and p.Arg229Gln. Finally, a patient was diagnosed with two homozygous pathogenic variants: one causing congenital adrenal hyperplasia (non- IEI) and the other in RFXANK gene causing MHCII deficiency.
A total of 194 variants were identified in the patients which included 90 missense, 33 nonsense (stop gained), 29 frameshift deletion, 17 intronic, 10 exon deletion, 10 insertion, 4 in frame deletion, 1 insertion deletion variants. One hundred forty-one pathogenic or likely pathogenic variants (72.7%), 44 variants of uncertain significance (VUS) (22.7%), and 9 benign or likely benign variants (4.6%) were identified in genes which would explain patients’ phenotypes. The genetic analysis allowed the identification of novel variants in different genes, for example, novel variants were detected in the BTK gene (p.His362Leu and p.Lys175Ter), in the ITGB2 gene (p.Cys459Ter, p.Gly167Val, p.Gln218Ter), and in the ADA gene (p.His17Arg, p.Pro55_Thr57del) (Table 1).
Based on the IUIS classification, the most common IEI subgroups within our cohort were immunodeficiency affecting cellular and humoral immunity (n = 128, 44.6%) (SCID (n = 87, 30.3%) and combined immunodeficiency (CID) generally less profound than SCID (n = 41, 14.3%)); congenital defects in the phagocytes (n = 60, 20.9%); diseases of immune dysregulation (n = 39, 13.6%); CID with associated or syndromic features (n = 24, 8.4%); defects in intrinsic and innate immunity (n = 16, 5.6%); predominant antibody deficiencies (n = 13, 4.5%); auto-inflammatory disorders other than FMF (n = 6, 2.1%); and finally complement deficiency was the least common (n = 1, 0.4%); it is worth noting a pathogenic variant in THEMIS gene that is not present in the IUIS classification 2019 [4] nor in its update in 2021 [6] was identified in one patient (Fig. 4).

The frequencies of different IEI subgroups in the studied patients with confirmed genetic diagnosis according to the IUIS 2019 classification
Genetic diagnosis was performed for 87 patients with SCID immunophenotyping. In T-B-NK + SCID/Omenn syndrome phenotype without microcephaly, variants in RAG1/2 were detected in 41 patients from 38 different families. Thirty-nine patients were diagnosed by Sanger sequencing out of 47 patients screened (82.9%) and two patients were diagnosed by NGS; pathogenic variants were detected by NGS in DCLERIC gene in 5 patients from 4 families including one patient with atypical phenotype who was diagnosed at the age of 17 years old. Three patients with T-B-NK + phenotype with microcephaly were diagnosed by NGS: NHEJ1 variant was detected in 2 siblings from the same family and LIG4 in another patient. Meanwhile, in T-B-NK- phenotype, ADA variants were detected in 9 patients from 8 different families.
Regarding the T-B + NK- phenotype, variants in IL2RG were identified in 8 male patients and JAK3 variants in 12 patients from 11 families. While for T-B + NK + phenotype, variants in IL7RA were detected in 4 patients from 3 families, CD247, CD3E, and LAT variants in one patient each. In two atypical patients with T-B + NK + phenotype, pathogenic variants in RAG1 and RAG2 genes were detected by WES.
Genetic diagnosis was performed for 41 patients with CID generally less profound than SCID: DOCK8 variants were identified in 23 patients originating from 18 different families (3 of which had associated CARD9 variants, and one had associated AIRE variants). Nine patients were diagnosed with MHCII deficiency having pathogenic variants in RFXANK in 5 patients from 4 families, RFX5 in 3 patients, and CIITA in 1 patient. Other genetic variants were identified for CID patients in DOCK2 gene in 3 patients from 2 families, one of them was previously reported [7]; CARD11 gene in 2 patients; Zap70, CD40L, and ARPC1B in 1 patient each.
Genetic diagnosis was done for 24 patients with CID with syndromic defects: WAS gene variants were detected in 6 patients from 5 different families; 4 patients with DiGeorge syndrome were confirmed by FISH. PNP variants were identified in 5 patients from 3 different families. Other rare genetic diagnosis including ATM, STAT3(AD LOF) were detected in 2 patients each while variants in PGM3, TCN2, IKBKG, ORAI-1, and IL6R were observed in one patient each.
Genetic diagnosis was done for 13 patients among the group with predominant antibody deficiencies. Pathogenic BTK variants were confirmed in 4 symptomatic male patients with defective BTK expression by flow cytometry while having normal B cell count and/or near normal immunoglobulin levels. Variants in AICDA gene was identified in 3 patients, and other rare variants in PIK3CD, NFKB1, NFKB2, CR2 were found in 1 patient each. WES in 2 unrelated patients revealed pathogenic/VUS variants in MSH6 gene associated with pathogenic variants in LRBA gene.
Genetic diagnosis was done for 39 patients presenting with immune dysregulation disorders. Twenty-one patients from 15 different consanguineous families had variants in LRBA gene (2 patients had associated MSH6 variants). Other less common genetic diagnosis found were variants in IL10RA and IL10RB in 4 patients; FOXP3 gene variants in 3 unrelated male patients; AIRE gene variants in 3 unrelated patients (in one of them an associated homozygous pathogenic DOCK8 gene variant was present); SLC7A7 gene variants in 3 patients from 2 different families; and UNC13D, PRKCD, SH2D1A, RIPK1, FAS variants were found in 1 patient each.
Genetic diagnosis was done for 60 patients with congenital defects of phagocyte number, function, or both. CGD was the most common phagocytic function defect in Egypt, Sanger sequencing for only 44 patients was done and revealed variants in CYBA in 22 patients from 19 different families, NCF1 in 13 patients from 9 different families, CYBB in 7 male patients from 6 families, NCF2 in 2 patients. LAD I was the second common functional defect and Sanger sequencing for 10 patients confirmed the presence of pathogenic variants in ITGB2 gene. While for patients with defects in the phagocytic counts, NGS/WES revealed variants in ELANE gene in 3 patients, JANGN1 variants in 2 siblings from the same family, and CLPB in 1 patient.
Genetic diagnosis was done for 16 patients with defects in the intrinsic or innate immunity. Among patients with chronic mucocutaneous candidiasis phenotype, gain-of-function variants in STAT1 gene were identified in 4 patients. CARD9 variants were detected in 3 patients; all of them had associated pathogenic DOCK8 variants, while for patients with a clinical suspension of Mendelian susceptibility to mycobacterial disease, 3 variants in IL12RB1 were found in 6 patients from 6 different families, a homozygous variant in IFNGR1 gene causing complete deficiency in 1 patients, a homozygous variant in IFNGR2 gene causing partial deficiency in 1 patients, and a variant in STAT2 in 1 patient.
Genetic diagnosis was done for 6 patients with autoinflammatory disorders other than FMF. A pathogenic variant was detected in PLCG2 gene in 1 patient together with an associated pathogenic variant in HUWE1 gene. Variants in each of NLRP3, NLRP12, STING1, PSTPIP1, SH3BP2 genes were found in 1 patient each. A summary of genetic results in relation to patients’ phenotypes and preliminary laboratory results is presented in Table 2.
Prenatal diagnosis (PND) was offered to 23 families in 35 pregnancies. Fetuses were diagnosed with homozygous pathogenic variants in 12 occasions (34.3%), wild genotype in 6 occasions (17.1%), and heterozygous pathogenic variants in 17 occasions (48.6%). When a heterozygous state was found (as an AR variant was tested), the maternal contamination of the samples was ruled out by means of comparing maternal human leukocyte antigen (HLA) typing with that of the chorionic villous samples. The pregnancies with normal or carrier fetuses were continued, while those with diseased fetuses were dealt with according to the families’ decisions after proper counselling (Fig. 5).

Bar chart representing the different families with known molecular diagnosis of IEI who asked for prenatal diagnosis service and the genetic result of the examined fetuses
Discussion
The genetic heterogeneity of the IEI as well as the delay in the diagnosis in atypical cases leads to significant morbidity and mortality. Establishing definitive genetic diagnosis is very important for patients’ management [8].
In 2016, Cairo University PID center had published its own 5 years of experience (2010–2014) in this field being one of the largest tertiary referral centers that receive patients from Egypt as well as from nearby Arab countries. The study helped to identify the spectrum and different patterns of IEI in Egyptian children; however, the genetic diagnosis was only available for 102 patients (22.26%) of the clinically diagnosed patients (n = 476) [9]. With the increased utilization of genetic testing and widening of the collaborative efforts with different centers, we were able to reach the genetic diagnosis for 778 patients out of 1496 patients diagnosed and followed up till 2021 (52%). It is worth noting that due to the increased experience with flow cytometry in our center, a definitive diagnosis was reached for many IEI phenotypes without the need for molecular testing for an additional number of patients 385/1496 (25.7%). The functional tests using flow cytometry in our center had been validated by the results of genetic testing in previous studies. This is very evident in CGD, LAD I, WAS, BTK, DOCK8, MHC II, and LRBA deficiency patients; thus, the need for genetics in these patients is limited to families who request prenatal diagnosis in future pregnancies [10,11,12,13]. As expected for a highly consanguineous population, most patients suffered from AR IEI disorders (82.6%), whereas XL recessive and AD modes of inheritance were less frequently encountered. This agrees with previous studies conducted on similar populations from Middle East and North Africa (MENA) [14], and disagrees with a study on 160 Chinese patients utilizing targeted NGS were the diagnostic yield was 43.8% with majority of patients having X-linked disease [15].
In our cohort, the most common IEI subgroups were immunodeficiency affecting cellular and humoral immunity (44.6%) followed by congenital defects in the phagocytes (20.9%), diseases of immune dysregulation (13.6%), CID with associated or syndromic features (8.4%), while defects in intrinsic and innate immunity and predominantly antibody deficiencies were much less common. In a recent study published about IEI from MENA region, the genetic approach was performing targeted genetic sequencing based on the phenotype. WES was done for patients in whom targeted sequencing was not diagnostic or if their clinical phenotypes resemble several genetic defects. Their diagnostic yield was 83% and the highest diagnosis was predominantly antibody deficiency [14].
In contrast to our findings, a study presenting one of the largest molecular studies from India showed that defects in intrinsic and innate immunity, diseases of immune dysregulation, and antibody deficiencies were the most common PIDs in their studied cohort. Their diagnostic yield was 42%, less than ours, and their molecular testing was done by Sanger sequencing and NGS targeting a customized panel [3].
WES testing of 350 PID Iranian patients revealed pathogenic/likely pathogenic variants in 35% of the cases, a percent higher than the results we obtained from our WES analysis although our overall yield is higher (55.2%) [16]. This is probably because of the genetic approach adopted in our center in which WES is done only in cases where targeted sequencing fails to reach the diagnosis.
In a study about the efficacy of NGS versus WES on a large PID cohort, Platt and his colleagues had a diagnostic yield close to ours (56%), yet they concluded that WES has advantage of lower cost than NGS. However, this approach can not be applied in many countries with limited genetic facilities [17].
Seventy-one genes (15.5%) out of the IEI known genes were identified in Egyptian patients, which highlights the importance of discovering the genetic makeup for each disease phenotype in each country. One patient had pathogenic variant in THEMIS gene which is not present in the IUIS 2019 classification nor in its update in 2021; thus, more extensive functional studies for this gene are highly recommended.
Sanger sequencing was used to identify the genetic defects in 108 patients (38.3%) mainly when patients presented with classical phenotype and in genes with small number of exons. For example, in T-B-NK + SCID/Omenn syndrome phenotype without microcephaly (n = 47), Sanger sequencing of RAG1/2 genes identified the pathogenic variants in 39 patients (82.9%), thus limiting the need for NGS to almost less than one-fifth of the patients with these phenotypes.
In T-B + SCID patients (n = 47), WES helped in the diagnosis of 26 patients with variants in ILRG and JAK3 in 19 patients with T-B + phenotype. Based on these results, Sanger sequencing for JAK3/IL2RG (based on patient’s gender, family history, and immunophenotype) will help in diagnosis of nearly 73% of these patients thus decreasing the need for NGS/WES to no more than 27% of patients in this group [18].
The molecular diagnosis helped to identify variants that were repeatedly detected in several patients from different families in the same gene and even being detected mainly in certain geographic distribution within the country. Target screening for these hot spots first—when utilizing Sanger sequencing—saved time and decreased the cost [12]. For example in T-B-NK + SCID patients, Gly35Val variant in RAG2 gene had been identified in 11 patients coming from three governorates from North Egypt, while the p.Thr215Ile variant and p.Arg229Gln had been detected together both in homozygous form in 6 patients mostly coming from upper Egypt. In DOCK8 deficiency, p.Ser1711Ter and p.Phe1045LeufsTer2 were identified in 11 patients (47.8% of genetically diagnosed DOCK8 patients). In CGD/NCF1 patients, only one variant, p.Tyr26HisfsTer26, had been identified in all patients and in CGD/CYBA, one variant, p.Val99ProfsTer90, had been detected in almost all patients. These two variants were also the only ones reported by another study from upper Egypt [19].
The molecular diagnosis elaborates double genetic affection in two independent genes in a few patients (n = 8). Three patients had pathogenic DOCK8 and VUS CARD9 variants. Two had extensive fungal infections to which they were receiving antifungal treatment. The third was diagnosed early being screened as a sibling of an index case and was transplanted once diagnosed.
Two patients with pathogenic LRBA variants had associated variants (one pathogenic, one VUS) in MSH6 gene. Both patients were referred to a specialist for investigating the probability of having autosomal recessive mismatch repair cancer syndrome. These coincidences might need more extensive studies on the probable cause for this linkage and at times may require adjustment of therapeutic modalities for some of the patients.
One patient had pathogenic DOCK8 and 2 heterozygous AIRE variants; one was previously published as disease-causing variant [20] and the other was a frameshift variant (disease causing by mutation taster); the patient had low T regs by flow cytometry. The patient was kept under close supervision for autoimmune manifestations. One patient had RFXANK pathogenic variant associated with a pathogenic variant in CYP21A2 gene causing congenital adrenal hyperplasia; thus, a specialist was involved in the patient management plan. One patient had a pathogenic variant in PLCG2 gene together with pathogenic variant in HUWE1 gene; this patient had delayed mental and motor milestones.
In some cases, the molecular testing may reveal a genetic diagnosis that was not expected by the patient’s phenotype; for instance, two patients presented with classical T-B + SCID phenotype underwent WES; interestingly, the first patient showed a homozygous variant in RAG1 gene and the other showed a homozygous variant in RAG2 gene [21]. A male patient presented with autoimmune lymphoproliferative syndrome phenotype and had 20% CD4-CD8-TCRαβ + cells, WES revealed homozygous pathogenic variant in IL12RB1.
A global genetic sequencing pilot program offered by Jeffrey Modell Foundation to identify specific IEI defects leads to alteration in disease management in 40% of patients [22]. Examples for changing the treatment modality based on molecular diagnosis was a SCID male infant with T-B-NK- phenotype, the molecular diagnosis of adenosine deaminase deficiency allowed the clinician to instantly start polyethylene-glycol-conjugated bovine adenosine deaminase (PEG-ADA), and in the absence of an HLA matched sibling donor, the patient was treated with autologous hematopoietic stem cell gene therapy (HSC-GT) for the correction of his immunodeficiency [23].
Patients with VEO-IBD are usually subjected to a precision medicine approach to choose between stem cell transplantation, antibiotics, abatacept therapy, or other therapies [24]. One of our VEO-IBD patients was diagnosed by NGS with homozygous variant in IL10RA gene; once diagnosis was reached, the patient was subjected to SCT from a completely matched sibling.
It is important to note that the evaluation of different variants pathogenicity is critical to formulate clinically reliable results. Despite the advances in computing technology, this process cannot be fully automated and still requires clinical and expertise judgment [25]. In the current study, we reported on 9 benign/likely benign variants by Varsome, however predicted to be pathogenic by other computational analysis, previously published in patients with similar phenotypes [20, 26] and/or had minor allele frequency. This highlights the importance of functional validation of variants whenever possible.
Identification of the pathogenic variants allowed to offer genetic counselling service and PND for many families. In 2017, PND was done in 12 pregnancies from 10 different families with testing only available for 5 genes initially (RAG1, RAG2, NCF1, NCF2, IL10RB) [27]. But with the growing experience and the increase in the number of genes available for Sanger sequencing in our center (5 more genes were added: ADA, CYBA, RFXANK, IL10RA, ITGB2), PND was performed in 35 pregnancies from 23 families. This helped these families choose among different available options.
It is worth noting that outlining the incidence of IEI in Egyptian patients is the basis in understanding the disease phenotype and is a nidus for building up a national Egyptian registry.
Conclusion
Molecular diagnosis of IEI patients is highly important specially in atypical phenotypes as genetic heterogeneity of the diseases and the delay in diagnosis leads to high morbidity and mortality. It helps in offering genetic counseling for many families and taking the best therapeutic decisions for the patients. Studying the candidate genes in each disease phenotype may help to find a founder pathogenic variant, a hot gene exon, and linked genetic inheritance, and validate the results of other easier, less expensive diagnostic tests. In many developing countries, genetic testing cost is still high or even unavailable; thus, each center should follow its own algorithm to select the patients that benefit most from genetic testing and the families who need definitive diagnosis. Collaboration and networking with other centers help in achieving genetic diagnosis and adjusting personalized treatment for many patients. Finally, studying genetic background of IEI patients from consanguineous populations helps better understand the molecular immunopathogenesis of IEI diseases.
Data Availability
The authors confirm that data supporting the findings of the study are available in the article. Raw data were generated in Cairo University Specialized Children Hospital. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Grumach AS, Goudouris ES. Inborn errors of immunity: how to diagnose them? Jornal de Pediatria. 2021;26(97):84–90.
Latif AH, Tabassomi F, Abolhassani H, Hammarström L. Molecular diagnosis of primary immunodeficiency diseases in a developing country: Iran as an example. Expert Rev Clin Immunol. 2014;10(3):385–96.
Arunachalam AK, Maddali M, Aboobacker FN, Korula A, George B, Mathews V, et al. Primary immunodeficiencies in India: molecular diagnosis and the role of next-generation sequencing. J Clin Immunol. 2021;41(2):393–413.
Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;11:1–6.
https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J Clin Immunol. 2021;41(3):666–79.
Alosaimi MF, Shendi H, Beano A, Stafstrom K, El Hawary R, Meshaal S, et al. T-cell mitochondrial dysfunction and lymphopenia in DOCK2-deficient patients. J Allergy Clin Immunol. 2019;144(1):306–9.
Mat Ripen A, Chear CT, Baharin MF, Nallusamy R, Chan KC, Kassim A, et al. A single-center pilot study in Malaysia on the clinical utility of whole-exome sequencing for inborn errors of immunity. Clin Exp Immunol. 2021;206(2):119–28.
Galal N, Meshaal S, Elhawary R, Abd ElAziz D, Alkady R, Lotfy S, Eldash A, et al. Patterns of primary immunodeficiency disorders among a highly consanguineous population: Cairo University Pediatric Hospital’s 5-year experience. J Clin Immunol. 2016;36(7):649–55.
Meshaal SS, El Hawary RE, Eldash A, Grimbacher B, Camacho-Ordonez N, AbdElaziz DS, Galal NM, et al. Diagnosis of DOCK8 deficiency using flow cytometry biomarkers: an Egyptian center experience. Clin Immunol. 2018;1(195):36–44.
Meshaal S, El Hawary R, Adel R, Abd Elaziz D, Erfan A, Lotfy S, Hafez M, et al. Clinical phenotypes and immunological characteristics of 18 Egyptian LRBA deficiency patients. J Clin Immunol. 2020;40(6):820–32.
El Hawary R, Meshaal S, Deswarte C, Galal N, Abdelkawy M, Alkady R, AbdElaziz D, et al. Role of flow cytometry in the diagnosis of chronic granulomatous disease: the Egyptian experience. J Clin Immunol. 2016;36(6):610–8.
El Hawary RE, Mauracher AA, Meshaal SS, Eldash A, AbdElaziz DS, Alkady R, Lotfy S, et al. MHC-II deficiency among Egyptians novel mutations and unique phenotypes. J Allergy Clin Immunol In Practice. 2019;7(3):856–63.
Aghamohammadi A, Rezaei N, Yazdani R, Delavari S, Kutukculer N, Topyildiz E, Ozen A, et al. Consensus Middle East and North Africa Registry on inborn errors of immunity. J Clin Immunol. 2021;29:1–3.
Xia Y, He T, Luo Y, Li C, Lim CK, Abolhassani H, Yang J, et al. Targeted next-generation sequencing for genetic diagnosis of 160 patients with primary immunodeficiency in south China. Pediatr Allergy Immunol. 2018;29(8):863–72.
Sherkat R, Khoshnevisan R, Bahreini A, Shahrooe M. Enigmas of Primary immunodeficiency disorders genetic diagnosis in our region. J Allergy Clin Immunol. 2020;145(2):AB124.
Platt CD, Zaman F, Bainter W, Stafstrom K, Almutairi A, Reigle M, Weeks S, et al. Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J Allergy Clin Immunol. 2021;147(2):723–6.
El Hawary R, Meshaal S, Mauracher AA, Opitz L, Abd Elaziz D, Lotfy S, Eldash A, et al. Whole-exome sequencing of T-B+ severe combined immunodeficiency in Egyptian infants, JAK3 predominance and novel variants. Clin Exp Immunol. 2021;203(3):448–57.
El-Mokhtar MA, Salama EH, Fahmy EM, Mohamed ME. Clinical aspects of chronic granulomatous disease in upper Egypt. Immunol Invest. 2021;50(2–3):139–51.
Meloni A, Perniola R, Faà V, Corvaglia E, Cao A, Rosatelli MC. Delineation of the molecular defects in the AIRE gene in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients from Southern Italy. J Clin Endocrinol Metab. 2002;87(2):841–6.
Meshaal SS, El Hawary RE, Abd Elaziz DS, Eldash A, Alkady R, Lotfy S, Mauracher AA, et al. Phenotypical heterogeneity in RAG-deficient patients from a highly consanguineous population. Clin Exp Immunol. 2019;195(2):202–12.
Quinn J, Modell V, Holle J, Truty R, Aradhya S, Johnson B, et al. Jeffrey’s insights: Jeffrey Modell Foundation’s global genetic sequencing pilot program to identify specific primary immunodeficiency defects to optimize disease management and treatment. Immunol Res. 2020;68(3):126–34.
Tucci F, Calbi V, Barzaghi F, Migliavacca M, Ferrua F, Bernardo ME, et al. Successful treatment with ledipasvir/sofosbuvir in an infant with severe combined immunodeficiency caused by adenosine deaminase deficiency with HCV allowed gene therapy with strimvelis. Hepatology. 2018;68(6):2434–7.
Snapper SB. Very–early-onset inflammatory bowel disease. Gastroenterology & hepatology. 2015;11(8):554.
Chinn IK, Bostwick BL. The role of genomic approaches in diagnosis and management of primary immunodeficiency. Curr Opin Pediatr. 2018;30(6):791–7.
Meshaal S, El Hawary R, Elsharkawy M, Mousa RK, Farid RJ, Abd Elaziz D, Alkady R, Galal N, et al. Mutations in Recombination Activating Gene 1 and 2 in patients with severe combined immunodeficiency disorders in Egypt. Clin Immunol. 2015;158(2):167–73.
El Hawary RE, Meshaal SS, Abd Elaziz DS, Elsharkawy MA, Alkady RS, Lotfy S, et al. Genetic counseling in primary immunodeficiency disorders: an emerging experience in Egypt. Mol Diagn Ther. 2017;21(6):677–84.
Acknowledgements
We would like to thank the patients and their families for their participation. We would also thank all collaborating centers that helped in the genetic diagnosis of the patients including the following: Laboratory of Human Genetics of Infectious Diseases (Necker Branch, France); Division of Immunology, Boston Children’s Hospital (Harvard Medical School, USA); Molecular Genetics Laboratory, Centre for Cardiovascular Surgery and Transplantation (Brno, Czech Republic); Division of Immunology, University Children’s Hospital Zurich (Zurich); Center for Chronic Immunodeficiency, University Medical Center Freiburg (Germany); Department of Immunology, Erasmus MC, University Medical Center Rotterdam (Netherlands); Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (Wien, Austria); Kinderklinik and Kinderpoliklinik IM DR. V. Haunerschen Kinder Hospital (Munich); and the Jeffrey Modell Foundation through their genetic sequencing pilot program.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation were performed by D. Abd Elaziz, R. Alkady, S. Lotfy, E. Chohayeb, M. Saad, J. Boutros, N. Galal, A. Elmarsafy. Data collection and analysis were performed by S. Meshaal, R. El Hawary, A. Eldash, R. Darwish, A. Erfan. The first draft of the manuscript was written by R. El hawary and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by Cairo University Faculty of Medicine Research Ethics Committee (N-114–2018).
Consent to Participate
Written informed consent was obtained from the parents of the patients.
Consent to Publish
Informed consents were obtained from patients’ guardians. The manuscript does not include any patients’ pictures or data that may identify them.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rabab E. El Hawary and Safa S. Meshaal contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
EL Hawary, R.E., Meshaal, S.S., Abd Elaziz, D.S. et al. Genetic Testing in Egyptian Patients with Inborn Errors of Immunity: a Single-Center Experience. J Clin Immunol 42, 1051–1070 (2022). https://doi.org/10.1007/s10875-022-01272-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01272-y