
Abstract
Background
Immunoglobulin A nephritis (IgAN) is the most common primary glomerulonephritis worldwide. It is caused by accumulation of IgA1-containing immune complexes in the kidney resulting in renal failure, which is thought to be due to altered glycosylation of IgA with a decrease of 2–3-sialylated galactosides (NeuAcα2-3Gal).
Purpose
The purpose of this study was to analyze whether altered glycosylation of IgA would lead to an altered binding to galectin-8, an endogenous lectin with strong affinity for 2–3-sialylated galactosides. Galectins are a family of β-galactoside-binding proteins; by binding various glycoproteins, they play important roles in the regulation of cellular functions in inflammation and immunity. Hence, an altered binding of IgA to galectin-8 could lead to pathologic immune functions, such as glomerulonephritis.
Methods
Affinity chromatography of serum glycoproteins on the human sialogalactoside-binding lectin galectin-8N permitted quantitation of bound and unbound fractions, including IgA.
Results
Analysis of ∼100 IgA nephritis sera showed that the galectin-8N unbound fraction of IgA increased compared to ∼100 controls, consistent with the known loss of galactosylation. A subgroup of ∼15% of the IgAN patients had a ratio of galectin-8 bound/unbound IgA <0.09, not found for any of the controls. Unexpectedly, the galectin-8N-binding fraction of serum glycoproteins other than IgA increased in the sera of IgAN patients but not in controls, suggesting a previously unrecognized change in this disease.
Conclusion
This is the first study that relates a galectin, an endogenous lectin family, to IgA nephritis and thus should stimulate new avenues of research into the pathophysiology of the disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Immunoglobulin A nephritis (IgAN) was first described in 1968 by Berger and colleagues; in an immunofluorescence study of renal biopsies from an unknown form of glomerulonephritis, mesangial deposits with IgA as their main component were found [1]. Detection of such IgA deposits in renal biopsies is still the only means to diagnose what is now recognized to be the most common glomerulonephritis worldwide, affecting 1.3% of the population [2], with 20–40% developing renal end stage failure within 20 years [3]. Population studies showing familiar clustering and geographical variations [4–6] and genetic studies [2, 7, 8] suggest a strongly inherited predisposition for IgAN, although no causative mechanism has been identified.
Considerable evidence suggests that the IgA deposited in the kidney has originated from circulating undergalactosylated IgA1 complexes [9–12]. Each IgA1 heavy chain contains up to six O-linked glycans in the hinge region between the CH1 and CH2 domains (Fig. 1). In normal IgA1, complete core-1 O-glycans like NeuAcα2,3Galβ1-3GalNAc linked to Ser or Thr are common (top glycan in Fig. 1), whereas in IgA nephritis IgA1, the O-glycans tend to lack the galactose residue as in GalNAc-Ser(Thr) or NeuAcα2,6GalNAc-Ser(Thr) (two bottom glycans in Fig. 1). In IgA nephritis, IgA-producing B cells have a glycosyltransferase imbalance in which a β1,3 galactosyltransferase and/or its chaperone Cosmc is downregulated [13, 14], resulting in decreased formation of Galβ1-3GalNAc (second glycan in Fig. 1), and increased activity of a α2,6 sialyltransferase resulting in increased addition of NeuAcα2,6 to the GalNAc (bottom glycan of Fig. 1) that also prevents further addition of Galβ1,3; the absent galactose in turn prevents further addition of NeuAcα2,3. Genetic variants of the β1,3 galactosyltransferase (C1GalT1) and its chaperone Cosmc with decreased activity and the α2,6 sialyltransferase with increased activity [15] may contribute to this and thereby predispose to IgA nephritis, but there may also be other non-genetic mechanisms for the aberrant glycosylation of IgA.

Simplified schematic of pathological O-glycosylation changes of IgA1 in IgAN and predicted binding of galectin-8. The two top O-glycans NeuAcα2,3Galβ1,3GalNAcα and Galβ1,3GalNAc are predominant (30–40% each) in IgA1 of normal sera [35], whereas the bottom two, GalNAcα and Neuα2,6GalNAcα, increase in IgA1 from IgAN patients, due to decreased activity of the galactosyltransferase adding the Galβ1,3 (black circle in top two glycans) and increased activity of the sialyltransferase adding the NeuAcα2,6 (vertical diamond in bottom glycan) to GalNAc (open square). Galectin-8N binds the top O-glycan with high affinity, but not the others [24]. The NeuAcα2,6 sialyltransferase may also act on the top structure to make NeuAcα2,3Galβ1,3(NeuAcα2,6)GalNAc, but also this structure has strongly reduced affinity for galectin-8N [24]. Thus, galectin-8N binding of IgA1 is expected to decrease in IgAN
Evidence from many studies suggest that the abnormal O-glycosylation of IgA1 found in IgAN patients plays a central role in the pathogenesis of the disease [11, 14, 16, 17], but the mechanism remains unclear [18, 19]. Altered aggregation, altered interaction with mesangial cells in the kidney, altered binding to IgA receptors, and IgG-specific recognition for exposed GalNAc all have been suggested to contribute either to increased deposition of IgA-containing complexes in the kidney or decreased clearance in the liver. Altered interaction with a carbohydrate-binding protein, a lectin, would be a reasonable hypothesis to explain the changed function of undergalactosylated IgA. Snail lectins specifically binding the exposed GalNAc residues have been used diagnostically for detection in patients [20, 21], but there have been few studies on the relationship of IgA glycosylation and the binding of endogenous lectins that might affect its function.
Galectin-8 is a good candidate for such a relationship. Galectin-8 contains two canonical galectin carbohydrate recognition domains (CRD) joined by a linker [22]. Like other galectin CRDs, they bind β-galactosides, but the N-terminal CRD of galectin-8 (galectin-8N) has a particularly high affinity if the β-galactoside is 2–3-sialylated and also bound with a 1–3 linkage to the next sugar, as found in NeuAcα2,3Galβ1,3GalNAc (top glycan of Fig. 1) [23, 24], and is the only mammalian galectin with high affinity for IgA [25]. In addition, a NeuAcα2,6 attached to the GalNAc residue strongly reduces affinity for galectin-8N [24]. This predicts that the carbohydrate structural and enzymatic changes in IgA nephritis patients described in the previous paragraph should reduce the binding of IgA1 to galectin-8N. In addition to this, galectin-8 exerts several immunoregulatory functions, playing a critical role in shaping the immune response and regulating inflammation [26, 27]. Depending on the context, galectin-8 can provide both inhibitory and stimulatory effects on immune cell adhesion [28, 29], in addition to induction of immune cell growth and apoptosis [30, 31]. In contrast to cytokines, galectins do not bind specific individual receptors, but rather “sense” the glycocode expressed on the cell surface of a cell. The dual activities of galectin-8 may therefore reflect a controlled regulation of glycosylation, e.g., activation and differentiation of immune cells but could also be a consequence of disease-associated glycosylation changes, e.g., immune escape mechanisms in which cell surface glycans can be altered to avoid immune recognition. For instance, we have recently found that sera from metastatic breast cancer patients contain two to three times more galectin-1 ligands and that this glycosylation change may result in altered function relevant for the disease [32]. In relation to the present study, using an autoimmune model of rheumatoid arthritis, galectin-8 has been shown to be trapped in the inflamed joint prohibiting it from inducing apoptosis in inflammatory cells resulting in an enhanced inflammatory response [33]. In light of these considerations, analyzing galectin-8 ligands in sera from IgAN patients has a great potential to not only detect the known alterations in glycosylation of the disease but may also suggest a pathophysiologically relevant function since these ligands most likely will encounter this galectin in tissue cells.
Material and Methods
Production of Recombinant Galectins
Recombinant galectin-8N and the mutant Q47A were produced in Escherichia coli BL21 Star (Invitrogen) in 1-l Luria–Bertani medium and stirred at 200 rpm at 37°C overnight. The expression of galectin-8N was induced by isopropyl-β-d-1-thiogalactoside, and the culture was further stirred for 3 h. The bacteria were collected by centrifugation, and the pellet was then suspended in phosphate-buffered saline (PBS; pH 7.2) containing 4-mM β-mercaptoethanol and 2-mM EDTA (MEPBS) and sonicated on ice for 1 min, 12 times. After centrifugation, the protein containing supernatant was run on a lactosyl-sepharose column. Galectin-8N was then eluted with MEPBS containing 150-mM lactose. Lastly, lactose was eliminated from galectin-8N with Centricon® Plus-70 Centrifugal Filter Units (Millipore).
Serum Samples
Serum samples from 20 healthy volunteers (average age 45, ratio male/female 60:40), 100 IgAN patients (average age 41, ratio male/female 76:24), and 92 patients with other forms of glomerulonephritis (average age 55, ratio male/female 38:58) (selected as the first non-IgAN sample in the bio-bank taken after the respective IgAN sample) were collected and stored as previously described [25].
Patients and Controls
The patients and the controls in this study were all participants in a long-term prospective study of glomerular diseases conducted at the Department of Nephrology, Lund University Hospital, Sweden. Serum samples were taken at time of kidney biopsy. Presenting symptoms were most often hematuria. After approval by the ethical committee at Lund University (LU 47-02), we obtained written informed consent from patients with biopsy-proven IgAN, diagnosed between February 1992 and November 2003. The morphological diagnoses were established by evaluation of representative percutaneous renal biopsy specimens by both light microscopy and direct immunofluorescence. The diagnosis of IgAN was based on the finding of IgA as the dominant or co-dominant immunoglobulin in a mesangial distribution pattern.
Out of the 87 patients included in the cohort, 30 (ratio male/female 28:2) patients reached end stage renal disease (ESRD), 3 (ratio male/female 2:1) died, and 6 (ratio male/female 4:2) patients were lost from follow-up. All other patients were followed up to the last planned visit in 2009. The number, age, gender, and baseline data of patients are presented in Table I.
Neuraminidase Treatment of Sera
One-milliliter sera from healthy subject H2 were treated with 0.5 μmol of neuraminidase (sialidase) from Vibrio cholerae (Roche) in 10-mM sodium acetate buffer, pH = 5, for 1 h at 37°C.
Galectin Affinity Chromatography
Galectin-8N and galectin-8 Q47A were coupled to 1-ml NHS-activated Hi-Trap affinity columns (Amersham Biosciences) as described in [25]. Sample loading, washing, and elution were carried out as described previously [25], protein concentrations determined with the Bio-Rad protein assay, and fractions stored at −20°C until further analysis. Columns were used for approximately 20 chromatographed sera. Within this limit, there was <4% variation in the bound protein amount, when the same serum was rechromatographed, as shown for 12 different sera of IgAN patients and controls. For some sera, the unbound fraction was chromatographed a second time on the same reconstituted column, but no more proteins bound (not shown), demonstrating that the column was not overloaded, and bound proteins not overlooked because of competition with other proteins. In some cases, the gel material of finished columns was analyzed by SDS-PAGE for possible protein retention after lactose elution, as described previously [25].
SDS-PAGE
Serum ligands were analyzed by one-dimensional 4–20% SDS-PAGE, all as described previously [25].
IgA Measurements
IgA content of sera (diluted 1/10 in PBS) or pooled galectin-8N binding fractions from 98 IgAN patients, 20 healthy donors, and 76 controls with IgAN symptoms were analyzed with nephelometry, IMMAGE 800 (Beckman Coulter) using a low concentration Immunoglobulin A Reagent kit (Beckman Coulter).
LC–MS/MS
Four samples (H2, K2, P2 and P4) of galectin-8N bound serum glycoproteins were analyzed by LC–MS/MS of pooled tryptic peptides to identify major protein components and estimate their relative abundance as described in Electronic Supplementary Material—Supplementary methods.
Results
Binding of Serum Glycoproteins to Galectin-8N
The binding of serum glycoproteins to galectin-8N was analyzed by affinity chromatography (first trace in Fig. 2). This resulted in sharp separation between an unbound flow through fraction and a bound fraction that could be eluted with lactose. The main components of the bound fraction are given in Fig. 3a, as determined previously by MALDI-TOF MS and confirmed by western blotting [25].

Affinity chromatography of untreated or 2,3-neuraminidase-treated serum on immobilized human galectin-8N. Chromatograms of serum from a healthy individual treated with specific neuraminidase (NA) or untreated (UT) and subjected to affinity chromatography with immobilized galectin-8N or the galectin-8 Q47A mutant, deficient in binding to sialylated galactosides. The protein concentration of each fraction (0.2 or 1 ml) is given on the Y-axis. Elution with lactose (150 mM) started after washing with 32-ml PBS (arrow heads). Each chromatogram has been moved for clarity by +0.1 on the Y-axis and by +5 on the X-axis

Identification of major galectin-8N binding serum glycoproteins. a SDS-PAGE (4–20% stained with Coomassie) of galectin-8N bound glycoproteins with major proteins indicated as previously identified by Western blot and MALDI-TOF-MS [25]. b Estimated relative amounts of galectin-8N bound serum proteins in four samples by LC–MS/MS analysis of pooled tryptic peptides. The serum concentrations of galectin-8N bound total protein and IgA were determined by protein assay and nephelometry as given in Table SI. The relative amounts of the remaining galectin-8N bound proteins were determined by LC–MS/MS assuming a linear relationship between sample protein concentration and the summed abundances for the peptides uniquely mapping to each protein (Tables SII and SIII). c SDS-PAGE of galectin-8N unbound (UB) and bound (B) fractions with the highest protein concentrations from four healthy individuals, four IgAN patients, and three controls with other histological patterns of glomerulonephritis. Indicated to the left are the mobilities of known size markers. The major visible bands of the unbound fractions correspond to albumin (at 67 kDa), transferrin (above the albumin), and IgG heavy chain (below the albumin)
A deeper proteomics characterization by LC–MS/MS of pooled tryptic peptides from the bound fraction of four samples revealed over 100 proteins (Table SII). Their concentrations were estimated from the MS data and the percentages in the galectin-8N bound fraction by comparing with known total concentrations in serum (Table SIII). For some proteins, the percentage in the bound fraction was very low, and they are likely contaminants from the unbound fraction. Using albumin as a marker for this (0.3% in bound fraction) and giving a margin for the variability of MS signal for different protein (as discussed in Electronic Supplementary Material—Supplementary methods), we defined protein with <2% in bound fraction as non-binding, proteins >10% in the bound fraction as clear significant galectin-8N binders, and an intermediate group with 2–10% in the bound fraction as likely binders but at a low level.
The group of clear significant galectin-8N bound proteins (>10%) included as major components and in agreement with Fig. 3a, IgA1, haptoglobin, α-2-macroglobulin, and hemopexin (Fig. 3b), but also lower levels of inter-alpha-trypsin inhibitor, orosomucuid, and a few other proteins (Table SIII). These are included in the group “Others” in Fig. 3b, with low binders (2–10%) such as apolipoproteins, protease inhibitors, IgM, and some more. The non-binders (<2%) included among major serum glycoproteins, transferrin, complement C3, IgG, and IgA2. Thus, galectin-8N binding appears specific for IgA1 among immunoglobulins, with a possible lower binding to IgM. This is in contrast to, for example, galectin-1 where IgM is one of its major serum ligands [32].
The analysis of multiple bound fractions by SDS-PAGE (lanes marked B in Fig. 3c) agreed with the results described above for a wider range of samples, including the rough quantitation, for example, sample P2 shows high haptoglobin both in panels b and c of Fig. 3. The main visible components of unbound fractions (lanes marked UB in Fig. 3c) were albumin, IgG, and transferrin.
Galectin-8N did not bind any glycoproteins from serum treated with neuraminidase (second trace in Fig. 2). Moreover, the Q47A mutant of galectin-8, which has a decreased affinity for sialylated glycans [24, 34], bound much less serum glycoproteins (about 0.5 mg/ml) (third trace in Fig. 2). This shows that binding of galectin-8N to serum glycoproteins depends on the presence of 2–3-sialylated galactosides. This is in contrast to galectin-1 and galectin-3, for which neuraminidase treatment of serum gives increased binding (unpublished). The Q47A mutant bound slightly more glycoproteins from neuraminidase-treated serum (trace 4 in Fig. 2), which may be explained by its increased affinity for some non-sialylated galactosides [23, 24, 34].
Large Increase of Galectin-8N-Binding Serum Glycoproteins in IgAN Patients
Serum samples were collected from 192 patients at the time of kidney biopsy for the suspicion of glomerular disease. Of these, 100 had IgA deposits in the mesangium, indicating IgA nephritis, (P1–P100 in Supplementary Table I), and 92 patients (here designated “controls”) had other histological patterns of glomerulonephritis (K1–K92). In addition, sera from 20 age-matched healthy persons were included. All sera were fractionated by affinity chromatography on immobilized galectin-8N as described above (Fig. 2). The yield of galectin-8N-binding proteins from the IgAN sera was on average 3.3 mg/ml (range 0.3–7.3), whereas the yields were significantly (p < 0.0001, one-sample t test) lower for the sera of the controls (2.2 mg/ml; range 0.9–5.5) and healthy (2.1 mg/ml; range 0.8–4.1) (Fig. 4). Most of the increase of galectin-8N-binding proteins in IgAN sera was not due to IgA as described below, but in some cases due to haptoglobin (as for sample P2, Fig. 3b, c), and/or other glycoproteins.

Significant increase of serum proteins binding to galectin-8N in IgAN patients. Yields of galectin-8N binding serum glycoproteins (sum of amount in bound fractions multiplied by 10 to give milligram per milliliter of original serum) for sera from 100 IgAN patients, 20 healthy subjects, and 92 controls with IgAN symptoms. Horizontal lines mark the mean for each group (mean/median for each group (mg/ml sera): 3.3/3.1, 2.2/2.1, 2.1/2.1); the difference between IgAN patients and the two other groups was statistically significant (p < 0.0001) as calculated by one-way ANOVA. Yields from all sera are found in supplementary Table I
Significant Increase of Galectin-8 Non-binding IgA in IgAN Patients
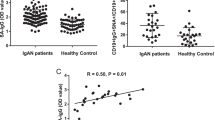
IgA levels were determined for all subjects, in unfractionated sera (total IgA) and in the galectin-8N bound fractions, and the difference gave galectin-8N unbound IgA (Fig. 5). Total IgA content in 98 IgAN patient sera was on average 3.4 mg/ml serum (range 1.5–7.7) compared to 2.3 (range 0.8–4.4) in 76 controls and 2.1 (range 0.1–4.3) in 17 healthy subjects (Fig. 5a). Most of this was due to galectin-8N non-binding IgA, on average 2.6 mg/ml (range 0.4–7.0) in IgAN compared to 1.6 (range 0.4–3.5) in controls and 1.5 mg (range 0.1–3.5) in healthy individuals (Fig. 5b). In contrast, there was only a small difference in the content of galectin-8N bound IgA between the subject groups (not shown). Plotting the ratio of galectin-8N bound/unbound IgA (Fig. 5c) revealed an interesting stratification of the IgAN patients. For the major group (patients), the ratio was similar as found for the controls and healthy, but a smaller group (16 patients) had a much lower ratio of galectin-8N bound/unbound IgA (<0.1), not found for any of the healthy and only one of the controls. A scatter plot of the ratio of galectin-8N bound/unbound IgA (<0.1) vs. total IgA again revealed that most IgAN patients fell in the same area as the healthy and controls (Fig. 5d). However, 15 IgAN patients clearly fell outside this because of the lower ratio and four additional ones because of higher total IgA.

Significant increase of galectin-8N non-binding IgA in sera from IgAN patients compared to healthy controls. a Total IgA (mean/median for each group (mg/ml sera): 3.4/3.3, 1,7/2.1, 2.3/2.4), b galectin-8N unbound (total bound) IgA (mean/median for each group (mg/ml sera): 2.6/2.4, 1.5/1.0, 1.6/1.7), and c ratio of galectin-8N bound/unbound serum IgA (logarithmic scale on the Y-axis) in sera from 98 IgAN patients, 17 healthy, and 76 kidney disease controls. d Scatter plot of ratio from c (Y-axis) vs. total IgA (X-axis). IgA in unfractionated serum (total) or galectin-8N bound fractions was quantitated using nephelometry. Horizontal lines mark the average for each group in a–c. The difference between groups was statistically significant (p < 0.0001) as calculated by one-way ANOVA
Relationship of Galectin-8N-Binding and Non-binding IgA to Disease Progression and Other Parameters in IgA Nephritis
When the time to end stage renal disease was plotted for 97 IgAN in a Kaplan–Meier type of graph (Fig. 6), there was a trend for a relationship of a low ratio of galectin-8N bound/unbound IgA (<0.5) to faster progress. However, the difference to the comparison group (ratio >0.5) was not significant (p ∼ 0.06), possibly because the follow-up time was somewhat limited for patients included in the last years of the study. Similar comparisons against total galectin-8N bound glycoprotein and total galectin-8N non-bound IgA did not give significant relationships to disease progression. The galectin-8-related parameters described here were also compared to a number of other patients’ parameters including serum initial and end creatinine, haptoglobin, classification of IgA deposits, age, and gender, but no significant correlations were found (not shown).

Indication of increased rate of disease progress to end stage renal disease for IgAN patients with low galectin-8 bound/unbound IgA ratio. Kaplan–Meier cumulative analysis of end stage renal disease for 87 IgAN patients, divided according to the ratio of galectin-8N bound/unbound IgA with a cutoff at a ratio 0.50 based on the mean in the control group. Patients with a ratio >0.50 may have better early (the first 9 years) renal survival than patients with a binding ratio <0.50, although statistical significance is slightly weaker than 0.05 level (p = 0.059, log rank test). Forty-one patients were lost from the follow-up group and are censored as indicated by vertical lines
Discussion
In the present study, we found that the IgA in patients with IgAN has reduced binding to galectin-8N and that this decrease is associated with disease severity. We demonstrate for the first time a clear difference in the binding ability of normal and altered IgA of IgAN to an endogenous lectin, galectin-8, suggesting a new type of functional difference related to the glycosylation. Unexpectedly, we also found that a higher proportion of other serum glycoproteins from IgAN patients bind galectin-8N.
The binding of IgA to galectin-8N as measured by affinity chromatography here requires a higher affinity than K d ∼5 μM [25]. Here we show that this binding is restricted to IgA1 and requires the presence of NeuAc; together with known specificity of galectin-8N, this indicates that the binding is mediated by the O-glycan NeuAcα2,3Galβ1,3GalNAc (the top glycan of Fig. 1), while the other O-glycans would not bind as described in the “Introduction”. Over 95% of the sialylated N-glycans would also not bind because the NeuAc is linked to the sixth position of Gal [35], which blocks binding of all galectins [22]. NeuAcα2,3Galβ1,3GalNAc makes up about 35% of the O-glycans on pooled healthy human serum IgA as estimated by [35]. Galectin-8N selectively binds a fraction of serum IgA (here on average 43% in healthy serum with the range 16–78%) containing this glycan, a feature that decreases in the altered IgA of IgAN.
In contrast, other investigators have measured features that increase in altered IgA in IgAN, mainly terminal GalNAcα (second last structure of Fig. 1), as detected by the binding of snail lectins [20, 36]. This gives a score for the lectin binding, but no quantitation of the proportion of IgA affected. The galectin-8N non-bound IgA measured here and the snail lectin-binding IgA measured in other studies probably represent overlapping fractions of the IgA, but may not be completely identical. The degree of partial segregation of IgAN patients from others based on both total IgA (Fig. 5a) and galectin-8N non-binding IgA (Fig. 5b) detected here is similar as found for total IgA [4, 37] and snail lectin-binding IgA in other studies [21, 36]. In addition, we identified a subgroup of the IgAN patients (about 15%) with particularly low ratio of galectin-8N bound/unbound IgA not found for any of the healthy and the kidney disease controls (Fig. 5c, d). In other studies, this subgroup would have been included among the subjects with increased GalNAcα (detected by snail lectin binding), but could not have been detected separately.
The sera studied here were collected at the time of renal biopsy, and there was no significant correlation with clinical outcome at that time point and the amount or proportion of unbound IgA. However, there was a trend for a correlation with subsequent progression to end stage renal disease and a low galectin-8N bound/unbound IgA ratio. Even though the significance was weak (p = 0.059, Fig. 6), the finding is intriguing and stimulates further studies.
The galectin-8N-binding fraction of serum glycoproteins other than IgA was unexpectedly significantly higher in the IgAN cases compared to the healthy and kidney disease controls (Fig. 4). This increase may be due to changes also in N-glycans as some of the proteins affected, e.g., haptoglobin, are not known to have O-glycans. Indeed, analyzing bound fractions from two IgAN patients with reversed phase LC–MS/MS revealed a large increase of bound haptoglobin in one of the patients (sample P2 in Fig. 3b, c). Although this protein is a likely candidate with known glycosylation changes in pathological conditions [32], the serum from other patients showed no increased levels of bound haptoglobin but possibly an increase of several other proteins, illustrating the complexity of the disease. One possible change is increased levels of 2–3-sialylated galactosides (NeuAcα2,3Galβ1-), a known but less common feature in normal human serum N-glycans, recognized to bind galectin-8N well [24]. This increase of galectin-8N-binding glycoproteins was neither seen for the kidney disease controls (Fig. 4) nor for 25 breast cancer patients [32], suggesting a relationship to IgAN. As for the changes in IgA described above, there was no significant correlation with particular disease parameters or progress of IgAN (not shown). Even if the binding sites are found only in a low percentage of N-glycans, they can result in binding of a high percentage of proteins carrying many glycans [32]. This may explain why the fraction of serum glycoproteins that bind galectin-8N is relatively large—on average 15% of all after exclusion of albumin, IgG, and IgA in healthy sera and on average 25% in sera from IgAN patients with some as high as 60%, suggesting that galectin-8 binding may represent a major pathophysiological change in the disease condition.
At present, it is only possible to speculate about the possible pathophysiological role of galectin-8 binding to IgA and other serum glycoproteins. It is not likely to have much effect in serum or plasma itself as there the concentration of galectins (low nanomolar range) is far below the concentration of the major bound glycoproteins. Instead, the functional role is likely when the glycoprotein encounters cells where the galectin is expressed. Galectin-8 has a wide expression in different cells [38], with particularly high expression in plasma cells [39] and lymphatic endothelial cells [40]. Moreover, high levels of galectin-8 have been found in synovial fluid cells of rheumatoid arthritis patients [33], and increased renal expression of other galectins has been found in other forms of nephritis [41, 42]. A number of possible cellular functions have been suggested for galectin-8. Roles in cell adhesion of Jurkat T cells and regulation of cell signaling by binding of glycoproteins have been proposed [43], as well as promoting differentiation of mature B cells into Ig-secreting plasma cells upon cell surface binding [39]. An emerging view is that galectins direct intracellular trafficking of glycoproteins in cells [44, 45], either after synthesis in the cell itself or after uptake by endocytosis as recently shown by us for haptoglobin and galectin-1 [32]. Thus, galectin-8 binding to IgA could act either while it is synthesized in plasma cells, or when it reassociates with cells after circulation, e.g., in the kidney mesangium where IgA-containing immune complexes deposit [46]. Recently, it has been shown that surface-attached galectin-8 is exposed on the membrane of thrombin-stimulated platelets [47] and that increased expression of factor V, an upstream signal of thrombin, causes the fibrin depositions connected to the development IgAN [48]. It is feasible that there will be an increased number of surface-exposed galectin-8 molecules in fibrinogenic mesangiolytic lesions that could act as a root for the formation of complexes. Our findings highlight the potential prospect and importance in using widely expressed endogenous lectins for detection of aberrantly glycosylated IgA in IgA nephritis and should stimulate further studies on the role of galectin-8 in the disease.
References
Berger J, Hinglais N. Intercapillary deposits of IgA–IgG. J Urol Nephrol (Paris). 1968;74(9):694–5.
Gharavi AG, Yan Y, Scolari F, Schena FP, Frasca GM, Ghiggeri GM, et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22–23. Nat Genet. 2000;26(3):354–7.
Novak J, Julian BA, Tomana M, Mesteck J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol. 2001;21(5):310–27.
Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19(5):1008–14.
Izzi C, Sanna-Cherchi S, Prati E, Belleri R, Remedio A, Tardanico R, et al. Familial aggregation of primary glomerulonephritis in an Italian population isolate: Valtrompia study. Kidney Int. 2006;69(6):1033–40.
Segelmark M, Hellmark T. Autoimmune kidney diseases. Autoimmun Rev. 2010;9(5):A366–71. doi:10.1016/j.autrev.2009.11.007.
Bisceglia L, Cerullo G, Forabosco P, Torres DD, Scolari F, Di Perna M, et al. Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Hum Genet. 2006;79(6):1130–4.
Paterson AD, Liu XQ, Wang K, Magistroni R, Song X, Kappel J, et al. Genome-wide linkage scan of a large family with IgA nephropathy localizes a novel susceptibility locus to chromosome 2q36. J Am Soc Nephrol. 2007;18(8):2408–15.
Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int. 2001;60(3):969–73.
Barratt J, Smith AC, Feehally J. The pathogenic role of IgA1 O-linked glycosylation in the pathogenesis of IgA nephropathy. Nephrology (Carlton). 2007;12(3):275–84.
Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN). Kidney Int. 2004;65(5):1544–7.
Czerkinsky C, Koopman WJ, Jackson S, Collins JE, Crago SS, Schrohenloher RE, et al. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J Clin Investig. 1986;77(6):1931–8.
Qin W, Zhou Q, Yang LC, Li Z, Su BH, Luo H, et al. Peripheral B lymphocyte beta1,3-galactosyltransferase and chaperone expression in immunoglobulin A nephropathy. J Intern Med. 2005;258(5):467–77.
Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Investig. 2008;118(2):629–39.
Zhu L, Tang W, Li G, Lv J, Ding J, Yu L, et al. Interaction between variants of two glycosyltransferase genes in IgA nephropathy. Kidney Int. 2009;76(2):190–8.
Allen AC, Harper SJ, Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin Exp Immunol. 1995;100(3):470–4.
Xu LX, Zhao MH. Aberrantly glycosylated serum IgA1 are closely associated with pathologic phenotypes of IgA nephropathy. Kidney Int. 2005;68(1):167–72.
Hiki Y. O-linked oligosaccharides of the IgA1 hinge region: roles of its aberrant structure in the occurrence and/or progression of IgA nephropathy. Clin Exp Nephrol. 2009;13(5):415–23.
Floege J. The pathogenesis of IgA nephropathy: what is new and how does it change therapeutic approaches? Am J Kidney Dis. 2011. doi:10.1053/j.ajkd.2011.05.033.
Gomes MM, Suzuki H, Brooks MT, Tomana M, Moldoveanu Z, Mestecky J, et al. Recognition of galactose-deficient O-glycans in the hinge region of IgA1 by N-acetylgalactosamine-specific snail lectins: a comparative binding study. Biochemistry. 2010;49(27):5671–82. doi:10.1021/bi9019498.
Oortwijn BD, Roos A, Royle L, van Gijlswijk-Janssen DJ, Faber-Krol MC, Eijgenraam JW, et al. Differential glycosylation of polymeric and monomeric IgA: a possible role in glomerular inflammation in IgA nephropathy. J Am Soc Nephrol. 2006;17(12):3529–39. doi:10.1681/ASN.2006040388.
Leffler H, Carlsson S, Hedlund M, Qian Y, Poirier F. Introduction to galectins. Glycoconj J. 2004;19(7–9):433–40.
Ideo H, Matsuzaka T, Nonaka T, Seko A, Yamashita K. Galectin-8-N-domain recognition mechanism for sialylated and sulfated glycans. J Biol Chem. 2011;286(13):11346–55. doi:10.1074/jbc.M110.195925.
Carlsson S, Oberg CT, Carlsson MC, Sundin A, Nilsson UJ, Smith D, et al. Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology. 2007;17(6):663–76.
Cederfur C, Salomonsson E, Nilsson J, Halim A, Oberg CT, Larson G, et al. Different affinity of galectins for human serum glycoproteins: galectin-3 binds many protease inhibitors and acute phase proteins. Glycobiology. 2008;18(5):384–94.
Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann N Y Acad Sci. 2010;1183:158–82. doi:10.1111/j.1749-6632.2009.05131.x.
Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin–glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9(5):338–52. doi:10.1038/nri2536.
Levy Y, Arbel-Goren R, Hadari YR, Eshhar S, Ronen D, Elhanany E, et al. Galectin-8 functions as a matricellular modulator of cell adhesion. J Biol Chem. 2001;276(33):31285–95. doi:10.1074/jbc.M100340200.
Carcamo C, Pardo E, Oyanadel C, Bravo-Zehnder M, Bull P, Caceres M, et al. Galectin-8 binds specific beta1 integrins and induces polarized spreading highlighted by asymmetric lamellipodia in Jurkat T cells. Exp Cell Res. 2006;312(4):374–86. doi:10.1016/j.yexcr.2005.10.025.
Hadari YR, Arbel-Goren R, Levy Y, Amsterdam A, Alon R, Zakut R, et al. Galectin-8 binding to integrins inhibits cell adhesion and induces apoptosis. J Cell Sci. 2000;113(Pt 13):2385–97.
Arbel-Goren R, Levy Y, Ronen D, Zick Y. Cyclin-dependent kinase inhibitors and JNK act as molecular switches, regulating the choice between growth arrest and apoptosis induced by galectin-8. J Biol Chem. 2005;280(19):19105–14.
Carlsson MC, Cederfur C, Schaar V, Balog CIA, Lepur A, Touret F, et al. Galectin-1-binding glycoforms of haptoglobin with altered intracellular trafficking, and increase in metastatic breast cancer patients. PLoS One. 2011;6(10):e26560.
Eshkar Sebban L, Ronen D, Levartovsky D, Elkayam O, Caspi D, Aamar S, et al. The involvement of CD44 and its novel ligand galectin-8 in apoptotic regulation of autoimmune inflammation. J Immunol. 2007;179(2):1225–35.
Carlsson S, Carlsson MC, Leffler H. Intracellular sorting of galectin-8 based on carbohydrate fine specificity. Glycobiology. 2007;17(9):906–12.
Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, et al. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions. J Biol Chem. 1998;273(4):2260–72.
Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71(11):1148–54. doi:10.1038/sj.ki.5002185.
Hashim OH, Shuib AS, Chua CT. Neuraminidase treatment abrogates the binding abnormality of IgA1 from IgA nephropathy patients and the differential charge distribution of its alpha-heavy chains. Nephron. 2001;89(4):422–5.
Lahm H, Andre S, Hoeflich A, Kaltner H, Siebert HC, Sordat B, et al. Tumor galectinology: insights into the complex network of a family of endogenous lectins. Glycoconj J. 2004;20(4):227–38. doi:10.1023/B:GLYC.0000025817.24297.17.
Tsai CM, Guan CH, Hsieh HW, Hsu TL, Tu Z, Wu KJ, et al. Galectin-1 and galectin-8 have redundant roles in promoting plasma cell formation. J Immunol. 2011;187(4):1643–52. doi:10.4049/jimmunol.1100297.
Cueni LN, Detmar M. Galectin-8 interacts with podoplanin and modulates lymphatic endothelial cell functions. Exp Cell Res. 2009;315(10):1715–23. doi:10.1016/j.yexcr.2009.02.021.
Ostalska-Nowicka D, Zachwieja J, Nowicki M, Kaczmarek E, Siwinska A, Witt M. Immunohistochemical detection of galectin-1 in renal biopsy specimens of children and its possible role in proteinuric glomerulopathies. Histopathology. 2007;51(4):468–76. doi:10.1111/j.1365-2559.2007.02818.x.
Kang EH, Moon KC, Lee EY, Lee YJ, Lee EB, Ahn C, et al. Renal expression of galectin-3 in systemic lupus erythematosus patients with nephritis. Lupus. 2009;18(1):22–8. doi:10.1177/0961203308094361.
Yamamoto H, Nishi N, Shoji H, Itoh A, Lu L-H, Hirashima M, et al. Induction of cell adhesion by galectin-8 and its target molecules in Jurkat T-cells. J Biochem. 2008;143(3):311–24. doi:10.1093/jb/mvm223.
Delacour D, Koch A, Jacob R. The role of galectins in protein trafficking. Traffic. 2009;10(10):1405–13. doi:10.1111/j.1600-0854.2009.00960.x.
Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129(1):123–34. doi:10.1016/j.cell.2007.01.049.
Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87.
Romaniuk MA, Tribulatti MV, Cattaneo V, Lapponi MJ, Molinas FC, Campetella O, et al. Human platelets express and are activated by galectin-8. Biochem J. 2010;432(3):535–47. doi:10.1042/BJ20100538.
Ono T, Liu N, Makino T, Nogaki F, Nomura K, Muso E, et al. Role of mesangial Factor V expression in crescent formation in rat experimental mesangioproliferative glomerulonephritis. J Pathol. 2004;204(2):229–38. doi:10.1002/path.1620.
Acknowledgements
We thank Barbro Kahl-Knutson and Christina Hansson for excellent technical support.
The work was supported by grants from the Swedish Research Council (Vetenskapsrådet) to HL and JM (project 2008-3356), from the Swedish Foundation for Swedish Research to JM (FFL4), from Swedish Healthcare System (ALF) to TH and MS, and from Region Skåne to HL.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
Supplementary methods and references (DOC 51 kb)
Table SII
Protein abundance signal from LC–MS/MS analysis of pooled tryptic peptides (MS1 signal) of galectin-8N bound samples from one healthy (H2), one control patient with other form of glomerulonephritis (K2), and two IgAN patients (P2 and P4) as described in Electronic Supplementary Material—Supplementary methods (PDF 14.3 kb)
Table SIII
Calculated concentration (microgram per milliliter serum) of proteins found in galectin-8N bound fractionsa,b (PDF 11.9 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Carlsson, M.C., Bakoush, O., Tengroth, L. et al. Galectin-8 in IgA Nephritis: Decreased Binding of IgA by Galectin-8 Affinity Chromatography and Associated Increased Binding in Non-IgA Serum Glycoproteins. J Clin Immunol 32, 246–255 (2012). https://doi.org/10.1007/s10875-011-9618-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-011-9618-3