
Abstract
Introduction
Spinal cord injury (SCI) is caused by two related but mechanistically distinct events: the primary injury to the spinal cord is caused by a mechanic trauma; the secondary injury is a cascade of cellular and molecular events that exacerbates the initial damage.
Materials and Methods
Neuroinflammation, an important event in the secondary injury cascade, is critical in the clearance of cellular debris after SCI. However, leukocytes and microglia, recruited to the injury site during neuroinflammation, can exacerbate the initial damage following SCI by secreting reactive oxygen species, matrix-metalloproteinase, and proinflammatory cytokines. Therefore, attenuating the activity of leukocytes and microglia is an attractive therapeutic strategy to reduce the neurological deficit associated with SCI.
Discussion
In this regard, immunoglobulin G (IgG) is a potential treatment candidate. IgG has been used clinically to treat autoimmune disease and has been demonstrated to attenuate the activities of leukocytes and microglia. In this review, we discuss the potential use of IgG for SCI based on the current understanding of the immune-modulating mechanism of IgG and the role of neuroinflammation in SCI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pathobiology of Spinal Cord Injury
According to the Christopher and Dana Reeve Foundation, there are over 1.2 million people living with spinal cord injury (SCI) in the United States alone [1]. Approximately 12,000 new cases in the USA are reported each year, and the average medical cost for a patient with SCI can be up to $10 million. Aside from the financial burden, people with SCI and their families and caregivers also deal daily with the physical, emotional, and social effects of this devastating condition. Few treatments are currently available or are even being investigated in clinical trials that address neurological impairment following traumatic SCI [2]. Therefore, there is a dire need for effective treatments that can reduce neurological deficit and improve a patient’s quality of life following SCI.
SCI is caused by two mechanistically and chronologically distinct events: the primary injury is caused by a mechanical force such as contusion, compression, or laceration, and the secondary injury is a cascade of cellular and molecular events that exacerbate the initial damage [3]. The secondary injury cascade includes the disruption of vasculature and the permeability of the blood-spinal cord barrier, edema, ionic toxicity, inflammation, cell deaths (necrosis and apoptosis), demyelination, and axonal degeneration (for a more extensive discussion, see [4]). While neural cells at the injury epicenter die quickly due to the primary injury, the cells in the surrounding regions of the epicenter are lost in a delayed fashion. The end result of the primary and secondary injury events is the substantial loss of neurons and oligodendrocytes, cavitation, and the formation of a glial scar that limits regeneration. Most research in the field has been focused on understanding the pathophysiology of the secondary damage and reducing the amount of delayed cell loss following SCI.
Role of Inflammation in SCI
Cellular and Molecular Response Following SCI
Inflammation, a key event in the secondary injury cascade, occurs immediately and persists for several weeks or months following SCI. The inflammatory response is critical for the clearance of cellular debris, which can prevent the regeneration of surviving neurons. However, overactivation of the inflammatory response can damage healthy tissue and exacerbate the damage, which can lead to poor functional recovery following SCI (for a more extensive discussion, see [4, 5]). Microglia are recruited to the injury site almost immediately. These cells secrete proinflammatory cytokines, including interleukin (IL)-1β, interleukin-6, and tumor necrosis factor-α (TNF-α), all of which increase the extent of inflammation.
The maintenance of proinflammatory cytokine expression is then accomplished by immune reactive cells, which are recruited to the injury epicenter. Endothelial cells upregulate their cellular adhesion molecules (CAMs) in response to the increased cytokines concentration, allowing leukocytes to adhere to the wall of endothelial cells and infiltrate the injury epicenter. The presence of neutrophils at the injury site peaks at 24 h, and they are cleared within 48 h. The presence of monocytes and lymphocytes is not observed at the injury site until 3 to 7 days after SCI.
Recruited Leukocytes and Microglia can Exacerbate SCI
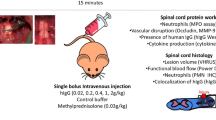
Blood-borne leukocytes and central nervous system (CNS) resident microglia/macrophages are recruited to the injury epicenter as part of the inflammatory response. These cells are essential in the clearance of cellular debris and in the promotion of cellular regeneration by the secreting of trophic factors and anti-inflammatory cytokines. However, in the acute phase of SCI, immune reactive cells can damage healthy tissues by releasing proinflammatory cytokines, reactive oxygen species (ROS), and matrix-metalloproteinase (MMPs, Fig. 1). Neutrophils can further increase the extent of the inflammatory response by producing proinflammatory mediators such as TNF-α, IL-1, and IL-8 [6]. MMP-9 and MMP-2 produced by neutrophils, macrophages, and endothelial cells can further break down the blood-brain barrier and increase leukocyte infiltration [7]. The influx of neutrophils and hematogenous macrophages, together with microglia, is a major source of ROS and inducible nitrous oxide synthase [8, 9]. These events can cause an increase in reactive oxygen radicals and nitrous oxide (NO) at the injury epicenter. Reactive oxygen radicals can react with NO to produce peroxynitrite (ONOO−) following injury [10]. The magnitude of the secondary damage can increase due to oxidation and nitrosylation of proteins, DNA, and lipids by reactive oxygen radicals and ONOO−. In addition, activated macrophages can physically induce axonal retraction and impede axonal regeneration [11].

Potential interactions of IgG with the neuroinflammatory response following SCI. Schematic diagram represents how the infiltration of microglia and leukocytes could exacerbate the initial damage. Microglia and leukocytes are recruited to the injury epicenter where they produce neurotoxic substances that cause neuronal and oligodendrocytic cell death in the secondary injury cascade. IgG has the potential to reduce the extent of secondary damage by interacting with microglia and leukocytes in the following mechanism: (1) inhibit microglia activation and reduce proinflammatory cytokine production, (2) inhibit leukocyte recruitment to the injury epicenter following SCI by acting on endothelial cells, and (3) inhibit leukocyte activation by upregulation of the inhibitory FcγIIB receptor. BBB blood-brain barrier, LIF leukemia inhibitory factor, ROS reactive oxygen species, NOS nitrous oxide synthase, MMPs matrix-metalloproteinases
Inflammation Following SCI: A Double-Edged Sword
Despite intensive research, the role of the inflammatory response in SCI is not fully understood. Many studies suggest both anti-inflammatory and proinflammatory treatments in SCI promote favorable outcomes. Anti-inflammatory treatments, such as the depletion of macrophages [12], the inhibition of MMP-9 [13], the reduction in the availability of CAMs [14], and the inhibition of neutrophil infiltration [15], are beneficial for recovery after SCI in animals and humans. The transplantation of macrophages into the injury site has been demonstrated to be beneficial to the recovery from SCI. In addition, wild-type mice were demonstrated to recover from SCI better than immune-deficient mice [16, 17]. Despite these studies, more research is needed to better understand the inflammatory process of spinal injury and to elucidate the exact benefits of proinflammatory and anti-inflammatory treatments.
Proposed Immunomodulating Mechanism of Intravenous Immunoglobulin G
Immunoglobulin G (IgG) isolated from pooled human serum has been used clinically to treat many autoimmune neuropathies such as Guillain–Barré syndrome. However, the mechanism underlying the observed benefits from intravenous IgG treatment is unclear. Many immunomodulating mechanisms for IgG have been proposed, and the exact one could potentially be a combination of many mechanisms. IgG preparations have been demonstrated to contain agonist anti-Fas antibodies, which induce monocyte and lymphocyte apoptosis via a caspase-dependent pathway [18]. IgG preparations also contain autoantibodies toward the sialic acid-binding immunoglobulin-like lectin-9 that can induce neutrophil apoptosis via caspase-dependent pathways and pathways dependent on ROS [19]. In addition, IgG has been demonstrated to inhibit the production of MMP-9 in cultured macrophages via its Fc and F(ab´)2 fragments [20]. IgG also has been demonstrated to bind neutrophil chemotactic factors C3a and C5a at low affinity via the constant region of the F(ab´)2 fragment [21]. C5a is a potent chemotactic factor for neutrophil and macrophage recruitment and activation [22]. Recently, it has been suggested that the IgG immunomodulating mechanism is achieved through the regulation of the expression levels of Fcγ receptors FcγRIIIA and FcγRIIB. [23] These receptors have low affinity for the Fc domain of the IgG molecules, and they are coexpressed on the surface of neutrophils, macrophages, mast cells, B-lymphocytes, and natural killer cells (for a detailed review, see [23]). These Fcγ receptors work antagonistically to maintain a constant balance between stimulatory and inhibitory signals in the immune system. The upregulation of the activating FcγRIIIA receptor has been linked to immune-complex diseases and autoimmune disorders, including Arthus reaction, rheumatoid arthritis, glomerulonephritis, systemic lupus erythematosus, and immune thrombocytopenic purpura [23]. Specifically, the sialylated N-linked glycan on the Fc fragment of IgG is required for the Fc fragment to bind to the SIGN-R1 (mice)/DC-SIGN (human) receptor on regulatory macrophages, which then upregulate the expression of immune inhibitory FcγRIIB receptors on effector macrophages [24]. The sialic acid residue is part of a glycan, which is linked to the Fc fragment at the asparagine at position 297.
IgG: A Potential Neuroprotective Treatment for SCI
IgG has been shown to modulate the immune response by inducing apoptosis in leukocytes, neutralizing components of the complement system, and inhibiting the activation of leukocytes. Therefore, using IgG to attenuate the detrimental effects of the inflammatory response in the acute phase of SCI is an attractive therapeutic strategy to reduce the neurological deficit associated with SCI and to improve patients’ functional recovery. IgG has been shown to reduce neurological deficit associated with CNS injury such as traumatic brain injury and stroke, both of which have similar pathobiology to SCI. To date, only one study presenting preliminary data on the use of IgG for SCI has been reported [25]. Therefore, extensive characterization of the potential neuroprotective property of IgG at the molecular, cellular, and behavioral level is required before moving IgG into clinical testing for SCI. Unpublished preliminary findings from our laboratory show that IgG treatment reduces secondary damage and improves hind-limb function following SCI in rats. This evidence provides a strong rationale to investigate the potential neuroprotective properties of IgG in greater detail and to fully elucidate the underlying mechanism for SCI.
References
Christopher and Dana Reeve Foundation. Paralysis facts and figures. http://www.christopherreeve.org/site/c.mtKZKgMWKwG/b.5184189/k.5587/Paralysis_Facts__Figures.htm. Accessed Nov. 25, 2009.
Baptise D, Fehlings M. Emerging drugs for spinal cord injury. Expert Opin Emerg Drugs. 2008;13(1):63–80.
Tator C, Fehlings M. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15–26.
Profyris C, Cheema S, Zang D, Azari M, Boyle K, Petratos S. Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol Dis. 2004;15(3):415–36.
Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and function recovery after spinal cord injury. Exp Neurol. 2008;209(2):378–88.
Cassatella M. The production of cytokines by polymorphonuclear neutrophils. Immunol. Today. 1995;16(1):21–6.
Fleming J, Norenberg M, Ramsay D, Dekaban G, Marcillo A, Saenz A, et al. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129(Pt 12):3249–69.
Clark R, Kochanek P, Schwarz M, Schiding J, Turner D, Chen M, et al. Inducible nitric oxide synthase expression in the cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr Res. 1996;39(5):784–90.
MacMicking JD, Xie Q, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–50.
Beckman J, Beckman T, Chen J, Marshall P, Freeman B. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–4.
Horn K, Busch S, Hawthorne A, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–41.
Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes B. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158(2):351–65.
Noble L, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22(17):7526–35.
Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, et al. Role of neutrophils in spinal cord injury in the rat. Neuroscience. 1997;79(4):1177–82.
Ditor DS, Bao F, Chen Y, Dekaban G, Weaver LC. A therapeutic time window for anti-CD 11d monoclonal antibody treatment yielding reduced secondary tissue damage and enhanced behavioral recovery following severe spinal cord injury. J Neurosurg Spine. 2006;5(4):343–52.
Hauben E, Agranov E, Gothilf A, Nevo U, Cohen A, Smirnov I, et al. Posttraumatic therapeutic vaccination with modified myelin self-antigen prevents complete paralysis while avoiding autoimmune disease. J Clin Invest. 2001;108(4):591–9.
Schori H, Shechter R, Shachar I, Schwartz M. Genetic manipulation of CD74 in mouse strains of different backgrounds can result in opposite responses to central nervous system injury. J Immunol. 2007;178(1):163–71.
Prasad N, Papoff G, Zeuner A, Bonnin E, Kazatchkine MD, Ruberti G, et al. Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J Immunol. 1998;161(7):3781–90.
von Gunten S, Schaub A, Vogel M, Stadler BM, Miescher S, Simon HU. Immunologic and functional evidence for anti-Siglec-9 autoantibodies in intravenous immunoglobulin preparations. Blood. 2006;108(13):4255–8.
Shapiro S, Shoenfeld Y, Gilburd B, Sobel E, Lahat N. Intravenous gamma globulin inhibits the production of matrix metalloperoteinase-9 in macrophages. Cancer. 2002;95(9):2032–7.
Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, et al. F(ab´) 2-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nat Med. 2003;9(4):431–8.
Marder S, Chenoweth D, Goldstein I, Perez H. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des. Arg J Immunol. 1985;134(5):3325–31.
Schmidt R, Gessner J. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100(1):56–7.
Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105(5):19571–8.
Gok B, Sciubba D, Okutan O, Beskonakli E, Palaoglu S, Erdarmar H, et al. Immunomodulation of acute experimental spinal cord injury with human immunoglobulin. G J Clin Neurosci. 2009;16(4):549–53. doi:10.106/j.jocn.2008.04.024.
Acknowledgments
The authors would like to thank Miss Allyson Tighe for her valuable comments and feedback. The authors would also like to acknowledge the Krembil Foundation and the PSI Foundation for the financial support. Dung Nguyen is currently funded by the Canadian Institute of Health Research Master’s Award.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fehlings, M.G., Nguyen, D.H. Immunoglobulin G: A Potential Treatment to Attenuate Neuroinflammation Following Spinal Cord Injury. J Clin Immunol 30 (Suppl 1), 109–112 (2010). https://doi.org/10.1007/s10875-010-9404-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-010-9404-7