
Abstract
Ocean emissions of inorganic and organic iodine compounds drive the biogeochemical cycle of iodine and produce reactive ozone-destroying iodine radicals that influence the oxidizing capacity of the atmosphere. Di-iodomethane (CH2I2) and chloro-iodomethane (CH2ICl) are the two most important organic iodine precursors in the marine boundary layer. Ship-borne measurements made during the TORERO (Tropical Ocean tRoposphere Exchange of Reactive halogens and Oxygenated VOC) field campaign in the east tropical Pacific Ocean in January/February 2012 revealed strong diurnal cycles of CH2I2 and CH2ICl in air and of CH2I2 in seawater. Both compounds are known to undergo rapid photolysis during the day, but models assume no night-time atmospheric losses. Surprisingly, the diurnal cycle of CH2I2 was lower in amplitude than that of CH2ICl, despite its faster photolysis rate. We speculate that night-time loss of CH2I2 occurs due to reaction with NO3 radicals. Indirect results from a laboratory study under ambient atmospheric boundary layer conditions indicate a k CH2I2+NO3 of ≤4 × 10−13 cm3 molecule−1 s−1; a previous kinetic study carried out at ≤100 Torr found k CH2I2+NO3 of 4 × 10−13 cm3 molecule−1 s−1. Using the 1-dimensional atmospheric THAMO model driven by sea-air fluxes calculated from the seawater and air measurements (averaging 1.8 +/− 0.8 nmol m−2 d−1 for CH2I2 and 3.7 +/− 0.8 nmol m−2 d−1 for CH2ICl), we show that the model overestimates night-time CH2I2 by >60 % but reaches good agreement with the measurements when the CH2I2 + NO3 reaction is included at 2–4 × 10−13 cm3 molecule−1 s−1. We conclude that the reaction has a significant effect on CH2I2 and helps reconcile observed and modeled concentrations. We recommend further direct measurements of this reaction under atmospheric conditions, including of product branching ratios.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The potential for tropospheric ozone destruction by iodine was recognized many decades ago (Chameides and Davis 1980) and confirmed later by measurements of the iodine oxide radical (IO) (Alicke et al. 1999; Read et al. 2008; Mahajan et al. 2010a, b; Gómez Martín et al. 2013; Großmann et al. 2013; Dix et al. 2013; Prados-Roman et al. 2015). A ubiquitous global marine boundary layer (MBL) background of around 0.5–1 pptv IO has been established by these measurements. The presence of IO establishes that active atmospheric iodine chemistry is occurring, driven by photochemical breakdown of iodine precursors. Iodine is a significant ozone-depleting agent in the MBL, being responsible for 20–30 % of the total ozone loss in that region (Saiz-Lopez et al. 2014; Prados-Roman et al. 2015; Sherwen et al. 2015). Calculations from global chemistry transport models indicate that globally iodine provides an odd oxygen (Ox) loss of 12–16 % of the total, at around ~720 Tg yr.−1 (Saiz-Lopez et al. 2014; Sherwen et al. 2015).
Oceanic emissions of the inorganic gases I2 and HOI, produced from ozonolysis of surface iodide (Carpenter et al. 2013; MacDonald et al. 2014), are believed to contribute ~2–2.5 Tg I yr.−1, around 80 % of global iodine emissions (Sherwen et al. 2015; Prados-Roman et al. 2015). However, in certain regions - including at high latitudes where surface ocean iodide levels are low, and in locations where wind speeds are high and surface ozone (O3(g)) mixing ratios are relatively low - organic gases may contribute 50 % or more of the iodine source (Prados-Roman et al. 2015). There are few oceanic flux measurements of CH2I2 and CH2ICl, but available data suggest that they contribute ~0.11 and 0.17 Tg I yr.−1 to the global atmosphere (Jones et al. 2010) similar in total to methyl iodide (CH3I) emissions (Carpenter et al. 2014).
Both CH2I2 and CH2ICl are known to have short photolysis lifetimes in the atmosphere of ~5 min and ~1 h at midday in mid-latitudes, respectively (Rattigan et al. 1997; Roehl et al. 1997; Mössinger et al. 1998), leading to strong diurnal cycles (Carpenter et al. 1999; Jones et al. 2010). Photolysis is assumed to be the sole driver of atmospheric loss of these compounds, and in the presence of chloride ions likely dominates aqueous loss also (Jones and Carpenter 2005; Martino et al. 2005). Laboratory determination of the photolysis rates in CH2I2 and CH2ICl in seawater indicate aqueous lifetimes at midday in mid- latitudes of 9–12 min for CH2I2 and 9–13 h for CH2ICl (Jones and Carpenter 2005; Martino et al. 2005).
So far, the possibility of night-time losses of the di-halomethanes remains unexplored in terms of analysis of field data and atmospheric modeling. Nakano et al. (2006) performed a kinetic study of the atmospheric reaction of NO3 with CH2I2 (reaction 1) and determined a rate constant of (4.0 ± 1.2) × 10−13 cm3 molecule−1 s−1. This would result in a nocturnal lifetime of ~3 h for CH2I2 in the clean MBL.
However this study was carried out at low pressure (25–100 Torr of N2 diluent) and there appear to be no corroborating studies in the literature.
Here we report seawater and air measurements and calculated ocean emissions of CH2I2 and CH2ICl from the eastern tropical Pacific Ocean. We focus on a 4-day period of near constant sea surface temperatures, chlorophyll a levels, and wind speeds, whilst sampling water from the North Equatorial Current. Measurements are compared to the 1D Tropospheric Halogen Chemistry Model (THAMO) (Saiz-Lopez et al. 2008) to test understanding of the atmospheric losses of CH2I2 and CH2ICl. We also infer a range of possible values for k CH2I2+NO3 from kinetic results obtained during a laboratory study of the reaction of the Criegee intermediate CH2OO with NO2¸which are the first data (to our knowledge) for this reaction obtained under ambient atmospheric boundary layer conditions (Ouyang et al. 2013).
2 Methods
2.1 Cruise track and shipboard measurements
The NOAA research vessel Ka’imimoana sailed from Pearl Harbour, Hawaii to Puntarenas, Costa Rica, via the 95oW and 110oW Tropical Atmosphere Ocean (TAO) buoy lines from 27th Jan. until 28th Feb. 2012 (Fig. 1). Very short-lived halocarbons (VSLH) were quantified in air and seawater at regular intervals during the cruise.

Cruise track of the R/V Ka’imimoana during the TORERO cruise during Jan./Feb. 2012, superimposed on chlorophyll-a background (MODIS) and showing wind speed and direction. The white rectangle shows the part of the cruise track chosen for the case study
2.1.1 Halocarbon measurements
Air was sampled from the top of the 10 m bow tower through a 60 m sampling line (1/2″ PFA, Swagelok). A diaphragm pump (KNF, NO35.1.2AN.18) was used to pull ~30 L min−1 down the line. At 1 m before the diaphragm pump, a T-union connected approximately 50 cm of 1/4″ heated instrument grade stainless steel line (Restek) to an inverted metal bellows pump (Senior Aerospace MB-158) which provided 10 psi of positive pressure to the instrument via ~1.5 m of 1/8″ heated instrument grade stainless steel tubing with a pressure relief valve maintaining flow and draining any water whilst the instrument was not sampling. An ozone analyser (Thermo Scientific Model 49i) drawing 3 L min−1 was also connected downstream of the metal bellows pump. Water was removed from the sample stream using a Nafion drier (Permapure, MD-050-72S-2) with a nitrogen counter current.
The absence of any line losses of VSLH using 100 m of 1/2″ PFA sampling line was verified in laboratory tests. A 35 L electropolished cylinder (Essex Cyrogenics) was filled with ambient air at 60 bar using a clean, high-pressure compressor (RIX SA-6). The cylinder air was (1) introduced directly into the instrument, using a pressure regulator and (2) used to over-flow the inlet of the pumped sampling system using 100 m of PFA line. This was performed at an open T-union so as not to influence the pressure in the sample line. The results of multiple analyses of air introduced directly and through the pumped sample line were compared and showed no significant losses (deviations less than the instrument reproducibility) for all of the halocarbons measured.
Calibrations were run daily using 1 L of a National Oceanic and Atmospheric Administration (NOAA) VSLH standard (SX-3570). All analytes in this standard were tested to be stable over 4 years by comparison with subsequently prepared standards, SX-3576 and SX-3581 at 2 and 4 years, respectively. In order to correct for short-term sensitivity changes between calibrations, atmospheric CCl4 was used as an internal standard. The air samples (1 L at 100 mL min−1), calibration and zero (nitrogen, n6.0) gas streams were selected (Markes, CIA8) and pre-concentrated on a cooled absorbent trap at −30 °C (Markes, Unity2). Samples were desorbed at 300 °C onto a gas chromatograph (GC, Agilent 7890) and separated on a capillary column (Restek, RTX502.2, 30 m, 0.25 mm ID, 1.25 μm) using an oven temperature ramp (40–250 °C, 3 min hold, 20 °C min−1 ramp). Eluents were ionized using electron ionization and analysed by an Agilent 5975C mass spectrometer (MS). CH2ICl and CH2I2 were measured using m/z 176/178 and 268/254 as quantification/qualification ions, respectively.
Surface seawater was sampled from the ship’s seawater inlet, located approx. 2–4 m (depending on sea-state) below the waterline, every 55 min (as well as at various depths from conductivity, temperature, depth (CTD) casts at specific points during the voyage, which are not discussed here). The seawater was pumped through the bow of the ship and through a vortex de-bubbler (Seabird) to remove any air bubbles, at a flow of around 10 L min−1 through ½” PFA tubing. A peristaltic pump was used to divert a portion of the water to an automated purge and trap (AutoP&T) system (Andrews et al. 2015). Briefly, the AutoP&T comprises a heated glass purge vessel automatically filled and emptied using a series of valves. A fixed sample volume (20 mL) was filtered (0.45 μm, PTFE filter) and de-gassed at 50 °C using 1 L of zero grade nitrogen at 50 mL min−1. Analysis was carried out using a second GC-MS with the same set-up as the air instrument. Similarly to the air measurements, atmospheric CCl4 was used to correct for short-term sensitivity changes between calibrations, by rapid sampling of 500 mL of air between each water sample.
2.1.2 Ozone measurements
Atmospheric ozone concentrations were measured throughout the cruise using a Thermo Scientific Model 49i Ozone Analyzer. The ozone analyzer was connected to the main air sampling line just downstream of the tee connecting the metal bellows pump.
2.2 Sea-air flux determination
Fluxes of VSLH across the ocean-atmosphere interface were calculated from the seawater-air concentration gradients using the two-layer model (Liss and Slater 1974);
and
where C w and C g are the respective bulk water and gaseous concentrations, H is the dimensionless gas- over-liquid form of the Henry’s law coefficient and k T , k w and k a are the total, liquid and air mass transfer coefficients, respectively. We use the Nightingale et al. (2000) parameterization for k w , and for k a the parameterization recommended by Johnson (2010). Pure water values of H from the compilation of Sander (1999) were corrected for salinity using the scheme of Johnson (2010).
2.3 THAMO model
The Tropospheric Halogen Chemistry model (THAMO) is a one-dimensional chemistry transport model with 200 stacked boxes at a vertical resolution of 5 m and a total height of 1 km (see Saiz-Lopez et al. 2008). The lowest level is the ocean surface, where gas phase deposition and upward flux can occur. Deposition to a specified aerosol surface area occurs with uptake coefficients taken from the recommendations of Sander et al. (2006) and Atkinson et al. (2000). The chemical scheme used for this study is from Saiz-Lopez et al. (2008) and is updated according to Mahajan et al. (2010a, b).
The modeled data discussed herein are the THAMO results at a height of 10 m above sea level (asl). The injection of CH2I2 and CH2ICl in the model is constrained by the fluxes measured in the field campaign. Photolysis rate constants for CH2I2 and CH2ICl were calculated by an explicit two-stream radiation scheme, which calculates the solar irradiance as a function of altitude, wavelength and solar zenith angle. For these simulations we assumed clear sky conditions. The modelled loss due to dry deposition of these species to the ocean is about 2 %. Measured O3 concentrations and assumed ranges of oceanic NO2 mixing ratios (see section 3.3) were used to calculate NO3 radical concentrations.
2.4 Laboratory measurements
Kinetics of the NO3 + CH2I2 reaction were derived from a laboratory study conceived primarily to investigate the reaction between stabilised Criegee intermediates (SCIs) and NO2. Full details are given in Ouyang et al. (2013); briefly, diiodomethane (CH2I2) was photolysed at 248 nm, in the presence of NO2 and a bath gas of synthetic air. 248 nm CH2I2 photolysis is known to lead to the formation of the CH2OO SCI in the presence of oxygen (Gravestock et al. 2010; Welz et al. 2012), and in turn form NO3 as a product of the CH2OO + NO2 reaction. NO3 formation was monitored by broad-band cavity enhanced absorption spectroscopy (BBCEAS), permitting insight into the CH2I2 + NO3 reaction. Experiments were performed across the accessible experimental conditions (in particular, with regard to precursor abundance), including monitoring NO3 abundance as a function of the CH2I2 concentration, whilst keeping all other aspects constant; these data allow us to constrain the rate constant k(NO3 + CH2I2).
Experiments were performed in an atmosphere of synthetic air at ambient (1 bar) pressure and 295 ± 5 K temperature, with 450 ppb NO2 and with the CH2I2 mixing ratio varied from 56 to 541 ppb. The reagent mixture was continuously flowed through the reaction cell, and photolysed by the 248 nm output from an Excimer laser, expanded and re-collimated to fill the reaction cell, operating at a total power of approximately 200 mJ pulse−1 (monitored by a calibrated power meter located after the reaction cell), and with a pulse repetition frequency of 0.1 Hz (ensuring total gas replenishment between pulses). The resulting NO3 formation was measured by BBCEAS, after a ca. 4 s transit time from the photolysis reactor to the absorption instrument. Owing to the presence of substantial concentrations of NO2, the majority of the NO3 was titrated into N2O5 during the 4 s transit period; the BBCEAS was therefore operated using a heated inlet and absorption cell, to thermally decompose N2O5 to NO3 + NO2. The quantity measured was therefore technically the sum of NO3 + N2O5, as described in the Results.
3 Results
3.1 Kinetic data
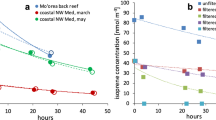
The measured variation in (NO3 + N2O5) with CH2I2 abundance is shown in Fig. 2. The observed behaviour was compared with the predictions of a simple model, which reproduced the evolution of NO3 between the photolysis pulse and the BBCEAS cavity (i.e. through the 4 s transit period), and which incorporated initial NO3 production, NO3/NO2/N2O5 equilibration, and the reaction of NO3 with CH2I2 to form (unreactive) products. Within the model, the NO2 and CH2I2 concentrations were fixed at their (known) values; standard NO2/NO3/N2O5 kinetics were taken from the MCMv3.1 (Bloss et al. 2005), and the initial NO3 abundance was calculated based upon the CH2I2 abundance, laser fluence and SCI yield; a single optimised value of the latter parameter was applied to all simulations. The rate constant for the NO3 + CH2I2 reaction, k 1, was varied, allowing the predicted variation of (NO3 + N2O5) with CH2I2 concentration to be compared with that observed as shown in Fig. 2. The experimental data clearly show an essentially linear dependence upon CH2I2, indicating that the NO3 + CH2I2 reaction has little effect under these (laboratory) conditions. The model data show that values of k 1 in excess of 5 × 10−13 molec cm−3 are not consistent with the observed behaviour, and we derive an upper limit of k 1 ≤ 4 × 10−13 molec cm−3, at 295 K and 1 bar total pressure (i.e. nominal ambient MBL conditions).

Measured (NO3 + N2O5) mixing ratio (filled grey circles, with ±2 σ error bars, reflecting combined precision and systematic (uncertainties in the NO3 absorption spectrum & fitting) components), compared with model predictions of the variation in (NO3 + N2O5) (coloured lines), as a function of CH2I2, for different values of k 1 (NO3 + CH2I2) in units of cm3 molecule−1 s−1
3.2 Field data
We choose as a case study 4 days of measurements during 29th Jan. – 2nd Feb. 2012 whilst transiting the North Equatorial Current in a southeasterly direction (Fig. 1). This period encountered stable surface chlorophyll a concentrations (0.1–0.2 μg m−3), sea surface temperatures (SSTs) (24.3 ± 0.3 °C), wind speeds (8.5 ± 0.8 ms−1) and wind direction (Fig. 1), and showed similar daily patterns of CH2I2 and CH2ICl atmospheric and seawater concentrations (Fig. 3).

Atmospheric mixing ratios, seawater concentrations, and sea-air fluxes of CH2I2 and CH2ICl measured during the TORERO cruise
Both gases displayed notable diurnal cycles in the atmosphere with mixing ratios steadily increasing during the night and rapidly decreasing at sunrise. Mean night-time/early morning maxima and day-time minima were (max: 0.047 +/− 0.01) pptv and (min: 0.023 +/− 0.004) pptv for CH2I2 and (max: 0.17 +/− 0.03) pptv and (min: 0.03 +/− 0.01) pptv for CH2ICl. Thus, the ratio of night-time maxima to day-time minima was more than a factor of two higher for CH2ICl mixing ratios (average of 5.3) than for CH2I2 (average of 2.0). This is surprising since the atmospheric photolysis lifetime of CH2I2 is much shorter than that of CH2ICl, as discussed in the Introduction. Seawater concentrations also showed diurnal cycles in CH2I2, as expected due to its fast photochemical decay in seawater (Jones and Carpenter 2005). Fluxes averaged 1.8 +/− 0.8 nmol m−2 d- 1 for CH2I2 and 3.7 +/− 0.8 nmol m−2 d- 1 for CH2ICl for the case study period, with night-time fluxes around 2–3 times higher than day-time for CH2I2 (but only 15 % higher and within the data variability for CH2ICl). Thus, in the absence of any night-time loss, nocturnal CH2I2 mixing ratios should be higher than expected given an assumption of a constant sea-air flux, whereas the reverse is true. Hourly fluxes are used to drive the 1D modeling, so such effects are taken care of in the analysis discussed in the next sections. Together, these results strongly suggest an additional night-time loss of CH2I2 in the atmosphere.
3.3 1D modelling
The THAMO model was used to investigate the effect of reaction (1) on the levels of observed CH2I2 during the TORERO campaign. Fig. 4 shows that there was no systematic difference between modeled and measured CH2ICl mixing ratios, suggesting that the model dynamics and mixing have the required level of skill, and that depositional losses are appropriate. Both the overall CH2ICl levels and the diurnal range are simulated well, and no night-time losses of CH2ICl are required to reconcile the model output with the observations.

Diurnal average CH2ICl mixing ratios during the period 29th Jan. – 2nd February 2012. Circles and black dotted lines show average of THAMO simulations. Blue crosses are hourly measurements, stripped of day of year. Blue line shows an hourly average of the measurements
Figure 5 shows however that for CH2I2, the base model (no NO3 reaction) overestimates night-time mixing ratios. On average over this period the model overestimation is a factor of 1.62. Figure 5 also shows the influence of including reaction (1) at different rate constants and NO2 (hence NO3 radical) concentrations.

The influence of different k 1 and NO2 mixing ratios on modelled CH2I2, compared with measurements. Lines are model results, purple triangles indicate CH2I2 measurements. Upper panel: 20 ppt NO2 at different k 1 (red - no reaction, magenta – k 1 = 5 × 10−14, green - k 1 = 2 × 10−13, dark blue - k 1 = 4 × 10−13 cm3 molecule−1 s−1); Middle panel: 30 ppt NO2 at different k 1 (lines as before); Lower panel: varying NO2 concentrations at k 1 = 2 × 10−13 cm3 molecule−1 s−1 (solid line - NO2 = 20 ppt, dashed line - NO2 = 30 ppt; dotted line - NO2 = 40 ppt)
Three out of the four days of data are simulated well with k 1 of 2 × 10−13 cm3 molecule−1 s−1 and 30–40 pptv NO2 (corresponding to 14–18 ppt peak NO3) or 4 × 10−13 cm3 molecule−1 s−1 and 20 pptv NO2 (corresponding to 9 ppt peak NO3). On the 31st January the model overestimates average CH2I2 night-time levels under all the scenarios considered here, although the maximum night-time concentration is simulated well under the highest loss scenario. Without additional measurements we cannot be conclusive on the reasons for this, although higher NO3 radical concentrations during this period are a possible explanation. Aircraft measurements made over the eastern Pacific Ocean as part of the NASA GTE/CITE 2 program indicate typical NO2 values of 18 +/− 15 ppt for maritime tropical air (Carroll et al. 1990) and ~50 ppt has been estimated as an upper limit for the remote ocean boundary layer from MAX-DOAS and satellite (SCIAMACHY and GOME-2) data (Peters et al. 2012).
4 Summary and conclusions
Our field data of atmospheric CH2I2 along with oceanic fluxes calculated from measured seawater concentrations are strongly indicative of a missing night-time sink of CH2I2, potentially through reaction with NO3 radicals. In the absence of NO3 measurements during our field study, it is not possible to infer the CH2I2 + NO3 rate constant with certainty, although a range of 2–4 × 10−13 cm3 molecule−1 s−1 offers generally good reconciliation between model and measurements. This range is consistent with a laboratory-derived upper limit, presented herein, and previous direct measurements made at low pressure. Given the importance of this reaction for understanding the atmospheric behavior of CH2I2, we recommend further laboratory measurements of the rate constant and the products of the reaction. Nakano et al. (2006) suggest that the possible products may be the compounds produced from H- transfer and I-transfer reactions:
The available thermodynamic data suggest that channel 1a is favourable but 1b is endothermic (Atkinson et al. 2004; Marshall et al. 2005; Sander et al. 2006), indicating that the reaction NO3 + CH2I2 represents a loss of NOx to HNO3. Further reactions of CHI2 and CH2I are uncertain, although in both cases I atoms are eventually produced which will form IO in the presence of O3. Oxidation of CH2I (by O2) leads to the Criegee intermediate CH2OO along with other products (Gravestock et al. 2010; Stone et al., 2013 and references therein). In locations with high CH2I2 emissions and sufficient NO3, such as polluted coastlines, the reaction may result in production of nighttime IO, or at least in modification of the diurnal profile of IO.
References
Alicke B., Hebestreit K., Stutz J., Platt U.: Iodine oxide in the marine boundary layer. Nature. 397, 572–573 (1999)
Andrews, SJ, Hackenberg, SC, Carpenter, LJ.: Technical note: a fully automated purge and trap GC-MS system for quantification of volatile organic compound (VOC) fluxes between the ocean and atmosphere. Ocean Sci. 11: pp. 1–9 (2015). doi:10.5194/os-11-1-2015
Atkinson R., Baulch D.L., Cox R.A., Crowley J.N., Hampson J., Hynes R.G., Jenkin M.E., Kerr J.A., Rossi M.J., Troe J.: Summary of evaluated kinetic and photochemical data for atmospheric chemistry. IUPAC. J. Phys. Chem. Ref. Data. 29, 167–266 (2000)
Atkinson R., Baulch D.L., Cox R.A., Crowley J.N., Hampson J., Hynes R.G., Jenkin M.E., Kerr J.A., Rossi M.J., Troe J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume I - gas phase reactions of ox, HOx, NOx and SOx species. Atmos. Chem. Phys. 4, 1461–1738 (2004)
Bloss, C, Wagner, V, Jenkin, ME, Wirtz, K, Wenger, J, Bloss, WJ, Lee, JD. Heard, DE, and Pilling MJ (2005) Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons. Atmos. Chem. Phys. 5: pp. 641–664.
Carpenter L.J., Sturges W.T., Penkett S.A., Liss P.S., Alicke B., Hebestreit K., Platt U.: Short-lived alkyl iodides and bromides at mace head, Ireland: links to biogenic sources and halogen oxide production. J. Geophys. Res. 104(D1), 1679–1689 (1999)
Carpenter L.J., MacDonald S.M., Shaw M.D., Kumar R., Saunders R.W., Parthipan R., Wilson J., Plane J.M.C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 6, 108–111 (2013). doi:10.1038/ngeo1687
Carpenter, LJ, Reimann, S, Burkholder, JB, Clerbaux, C, Hall, BD, Hossaini, R, Laube, JC, Yvon-Lewis, SA.: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol: Chapter 1 in Scientific Assessment of Ozone Depletion: 2014, Global ozone research and monitoring project – report no. 55, World Meteorological Organization, Geneva (2014)
Carroll M.A. et al.: Aircraft measurements of NOx over the eastern Pacific and continental United States and implications for ozone production. J. Geophys. Res. 95, 10,205–10,233 (1990)
Chameides W.L., Davis D.: Iodine: its possible role in tropospheric photochemistry. J. Geophys. Res. 85, 7383–7398 (1980)
Dix B., Baidar S., Bresch J.F., Hall S.R., Schmidt K.S., Wang S., Volkamer R.: Detection of iodine monoxide in the tropical free troposphere. P. Natl. Acad. Sci. USA. 110, 2035–2040 (2013). doi:10.1073/pnas.1212386110
Gómez Martín J.C., Mahajan A.S., Hay T.D., Prados-Román C., Ordóñez C., MacDonald S.M., Plane J.M.C., Sorribas M., Gil M., Paredes Mora J.F., Agama Reyes M.V., Oram D.E., Leedham E., Saiz-Lopez A.: Iodine chemistry in the eastern Pacific marine boundary layer. J. Geophys. Res.-Atmos. 118, 887–904 (2013). doi:10.1002/jgrd.50132
Gravestock T.J., Blitz M.A., Bloss W.J., Heard D.E.: A multidimensional study of the reaction CH2I + O2: products and atmospheric implications. Chem. Phys. Chem. 11, 3928–3941 (2010)
Großmann K., Frieß U., Peters E., Wittrock F., Lampel J., Yilmaz S., Tschritter J., Sommariva R., von Glasow R., Quack B., Krüger K., Pfeilsticker K., Platt U.: Iodine monoxide in the western Pacific marine boundary layer. Atmos. Chem. Phys. 13, 3363–3378 (2013). doi:10.5194/acp-13-3363-2013
Johnson M.T.: A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas. Ocean Sci. 6, 913–932 (2010)
Jones C.E., Carpenter L.J.: Solar photolysis of CH2I2, CH2ICl, and CH2IBr in water, saltwater, and seawater. Environ. Sci. Technol. 39, 6130–6137 (2005)
Jones, CE, Hornsby, KE, Sommariva, R, Dunk, RM, von Glasow, R, McFiggans, G, Carpenter, LJ.: Quantifying the contribution of marine organic gases to atmospheric iodine. Geophys. Res. Lett. 37: L18804 (2010).
Liss P.S., Slater P.G.: Flux of gases across the air-sea interface. Nature. 247, 181–184 (1974)
MacDonald S.M., Gómez Martín J.C., Chance R., Warriner S., Saiz-Lopez A., Carpenter L.J., Plane J.M.C.: A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modeling. Atmos. Chem. Phys. 14, 5841–5852 (2014)
Mahajan A.S., Plane J.M.C., Oetjen H., Mendes L., Saunders R.W., Saiz-Lopez A., Jones C.E., Carpenter L.J., McFiggans G.B.: Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic ocean. Atmos. Chem. Phys. 10, 4611–4624 (2010a). doi:10.5194/acp-10-4611-2010
Mahajan, AS, Shaw, M, Oetjen, H, Hornsby, KE, Carpenter, LJ, Kaleschke, L, Tian-Kunze, X, Lee, JD, Moller, SJ, Edwards, P, Commane, R, Ingham, T, Heard, DE, Plane, JMC.: Evidence of reactive iodine chemistry in the Arctic boundary layer. J. Geophys. Res., 115: D20303, (2010b). doi: 10.1029/2009JD013665
Marshall P., Srinivas G.N., Schwartz M.: A computational study of the thermochemistry of bromine- and iodine-containing methanes and methyl radicals. J. Phys. Chem. A. 109, 6371–6379 (2005)
Martino M., Liss P.S., Plane J.M.C.: The photolysis of dihalomethanes in surface seawater. Environ. Sci. Technol. 39(18), 7097–7101 (2005)
Mössinger J., Shallcross D.E., Cox R.A.: UV–VIS absorption cross-sections and atmospheric lifetimes of CH2Br2, CH2I2 and CH2BrI. J. Chem. Soc. Faraday Trans. 94, 1391–1396 (1998)
Nakano Y., Ukeguchi H., Ishiwata T.: Rate constant of the reaction of NO3 with CH2I2 measured with use of cavity ring-down spectroscopy. Chemical Physics Letters. 430, 235–239 (2006). doi:10.1016/j.cplett.2006.09.002
Nightingale P.D. et al.: In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Glob. Biogeochem. Cycles. 14, 373–387 (2000)
Ouyang B., McLeod M.W., Jones R.L., Bloss W.J.: NO3 radical production from the reaction between the criegee intermediate CH2OO and NO2. Phys. Chem. Chem. Phys. 15, 17070–17075 (2013)
Peters E., Wittrock F., Großmann K., Frieß U., Richter A., Burrows J.P.: Formaldehyde and nitrogen dioxide over the remote western Pacific Ocean: SCIAMACHY and GOME-2 validation using ship-based MAX-DOAS observations. Atmos. Chem. Phys. 12, 11179–11197 (2012). doi:10.5194/acp-12-11179-2012
Prados-Roman C., Cuevas C.A., Hay T., Fernandez R.P., Mahajan A.S., Royer S.J., Galí M., Simó R., Dachs J., Großmann K., Kinnison D.E., Lamarque J.F., Saiz-Lopez A.: Iodine oxide in the global marine boundary layer. Atmos. Chem. Phys. 15, 583–593 (2015)
Rattigan O.V., Shallcross D.E., Cox R.A.: UV absorption cross-sections and atmospheric photolysis rates of CF3I, CH3I, C2H5I and CH2ICl. J. Chem. Soc. Faraday Trans. 93, 2839–2846 (1997)
Read K.A., Mahajan A.S., Carpenter L.J., Evans M.J., Faria B.V.E., Heard D.E., Hopkins J.R., Lee J.D., Moller S., Lewis A.C., Mendes L., McQuaid J.B., Oetjen H., Saiz-Lopez A., Pilling M.J., Plane J.M.C.: Extensive halogen-mediated ozone destruction over the tropical Atlantic ocean. Nature. 453(7199), 1232–1235 (2008)
Roehl C.M., Burkholder J.B., Moortgat G.K., Ravishankara A.R., Crutzen P.J.: Temperature dependence of UV absorption cross sections and atmospheric implications of several alkyl iodides. J. Geophys. Res. 102, 12819–12829 (1997)
Saiz-Lopez A., Plane J.M.C., Mahajan A.S., Anderson P.S., Bauguitte S.J.B., Jones A.E., Roscoe H.K., Salmon R.A., Bloss W.J., Lee J.D., Heard D.E.: On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime. Atmos. Chem. Phys. 8, 887–900 (2008). doi:10.5194/acp-8-887-2008
Saiz-Lopez A., Fernandez R.P., Ordóñez C., Kinnison D.E., Gómez Martín J.C., Lamarque J.-F., Tilmes S.: Iodine chemistry in the troposphere and its effect on ozone. Atmos. Chem. Phys. 14, 13119–13143 (2014)
Sander, R.: Compilation of Henry’s law constants for inorganic and organic species of potential importance in environmental chemistry (version 3), http://www.mpchmainz.mpg.de/~sander/re/henry.html. (1999)
Sander S.P., Friedl R.R., Ravishankara A.R., Golden D.M., Kolb C.E., Kurylo M.J., Huie R.E., Orkin V.L., Molina M.J., Moortgart G.K., Finlayson-Pitts B.J.: Chemical kinetics and photochemical data for use in atmospheric studies, evaluation number 14. Administration, Jet Propulsion Laboratory and National Aeronautics and Space (2006)
Sherwen T., Evans M.J., Carpenter L. J., Andrews S. J., Lidster R. T., Dix B., Koenig T. K. , Volkamer R., Saiz-Lopez A., Prados-Roman C., Mahajan A. S. , Ordóñez C., Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys. Discuss. 15, 20957–21023 (2015)
Stone D., Blitz M., Daubney L., Ingham T., Seakins P.: CH2OO Criegee biradical yields following photolysis of CH2I2 in O2. Phys. Chem. Chem. Phys. 15, 19119–19124 (2013)
Welz O., Savee J.D., Osborn D.L., Vasu S.S., Percival C.J., Shallcross D.E., Taatjes C.A.: Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2. Science. 335, 204 (2012)
Acknowledgments
LJC acknowledges NERC (NE/J00619X/1) and the National Centre for Atmospheric Science (NCAS) for funding. The laboratory work was supported by the NERC React-SCI (NE/K005448/1) and RONOCO (NE/F005466/1) grants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Carpenter, L.J., Andrews, S.J., Lidster, R.T. et al. A nocturnal atmospheric loss of CH2I2 in the remote marine boundary layer. J Atmos Chem 74, 145–156 (2017). https://doi.org/10.1007/s10874-015-9320-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10874-015-9320-6