
Abstract
The crystal structure of 2′,3′,3′-tribromo-2′,3′-dihydrospiro[1,3-dioxolane-2,1′-indene], (C11H9Br3O2 ), has been determined by means of single-crystal X-ray diffraction methods. The title compound crystallizes in the monoclinic space group P21/n with unit cell parameters: a = 8.9799(4), b = 11.3368(8), c = 12.4233(9) Å, β = 94.938(4)°, V = 1260.04(14) Å3, Z = 4. The cyclopentane ring fused to the benzene ring adopts an envelope conformation with C11 at the tip of the envelope. The crystal structure is stabilized by C–H···Br and C–H···O interactions. The C atoms of the CH 2 groups of the 1,3-dioxolane ring, are disordered over two sites with an occupancy ratio of 0.62(7):0.38(7). A semiempirical quantum-mechanical calculation was carried out using the CNDO approximation.
Graphical Abstract
The crystal structure of 2′,3′,3′-tribromo-2′,3′-dihydrospiro[1,3-dioxolane-2,1′-indene], (C11H9Br3O2 ), has been determined by means of single-crystal X-ray diffraction methods. The title compound crystallizes in the monoclinic space group P21/n with unit cell parameters: a = 8.9799(4), b = 11.3368(8), c = 12.4233(9) Å, β = 94.938(4)°, V = 1260.04(14) Å3, Z = 4. The cyclopentane ring fused to the benzene ring adopts an envelope conformation with C11 at the tip of the envelope. The crystal structure is stabilized by C–H···Br and C–H···O interactions. The C atoms of the CH 2 groups of the 1,3-dioxolane ring, are disordered over two sites with an occupancy ratio of 0.62(7):0.38(7). A semiempirical quantum-mechanical calculation was carried out using the CNDO approximation.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Indanes are important class of molecules due to the pharmacological and medicinal properties [1–4] as well as natural product chemistry [5]. Brominations of hydrocarbons are important processes in synthetic chemistry [6–10]. The materials obtained from bromination of hydrocarbons have numerous industrial applications as pesticides, plastics, fire retardants and pharmaceutical chemicals [11]. Bromoindanes are important key intermediates in the industrial and laboratory preparation of hydroxy and epoxide compounds [12], and of indenone and fluorenone compounds [13].
In continuation of our investigations of the synthesis and structure of the bromoindanes, we describe herein the synthesis and structure of a new compound 2′,3′,3′-tribromo-2′,3′-dihydrospiro[1,3-dioxolane-2,1′-indene] (Scheme 1). In addition, a semiempirical quantum-mechanical calculation was carried out using the CNDO approximation. The theoretical CNDO and experimental X-ray structural results has been compared with each other.

Synthesis of 2′,3′,3′-tribromo-2′,3′-dihydrospiro[1,3-dioxolane-2,1′-indene]
Experimental
Synthesis
Melting points were determined on a Büchi model 530 apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on 400 (100) MHz spectrometers. Mass spectra (EI) were recorded at 70 eV as m/z. All solvents were dried and distilled before use. Column chromatography was performed on silica gel 60 (70–230 mesh, Merck). TLC was carried out on Merck 0.2 mm silica gel 60 F254 analytical aluminium plates. The substance reported in this paper is in its racemic form.
To a magnetically stirred solution of starting material 3′-bromospiro[1,3-dioxolane-2,1′-indene] (100 mg, 0.4 mmol) in dichloromethane (2 ml) at room temperature was added, dropwise, a solution of bromine (70 mg, 0,44 mmol) in dichloromethane (0.5 ml) over the course of 1 min. After stirring for 10 min at room temperature, the solvent was evaporated and the crude product, 160 mg (98 %) of tribromide, was crystallized from ether/hexane (3:1). m.p. 380–382 K.
1H NMR (400 MHz, CDCl3): 7.78 (d, J = 7.7 Hz, Hz), 7.55 (td, J = 1.0 Hz, J = 7.7 Hz, 2H), 7.42 (td, J = 1.0 Hz, J = 7.7 Hz, 2H), 7.32 (d, J = 7.7 Hz, 2H), 5.06 (s, 2H), 4.47 (m, 2H), 4.37 (m, 2H), 4.29 (m, 2H), 4.22 (m, 2H). 13C NMR (100 MHz, CDCl3): 145.0, 136.2, 131.6, 131.1, 125.8, 122.9, 110.9, 70.5, 66.83, 66.78, 59.6. IR (KBr, cm−1): 2956, 2895, 1465, 1316, 1273, 1193, 1149, 1090, 1052, 1027, 966, 948.
X-Ray Structural Analysis
Crystallographic data are given in Table 1. Pale yellow block crystal (0.2, 0.2, 0.2 mm) was used for data collection. Diffraction data for the title compound were collected at room temperature (T = 294(2) K) using a Rigaku R-AXIS RAPID-S diffractomer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data were corrected for Lorentz-polarization and absorption effects using CrystalClear [14]. The structure was solved by the direct method using SIR97 [15] and was refined by full matrix least squares based on F 2 using SHELXL-97 [16]. The molecular graphic were drawn using the ORTEP-3 for Windows [17] and PLATON programs [18].
H-atoms were positioned geometrically and refined using a riding model with C–H = 0.93 and 0.97 Å, and with Uiso(H) = 1.2 Ueq(C). All non-hydrogen atoms were refined anisotropically. Two poorly fitted reflections (4 9 0) and (7 2 0) were omitted from the refinement. The atoms, C1 and C2, of the –C–C– bond between the oxygen atoms in the 1,3-dioxolane ring, are disordered over two sites with site-occupancy factors of 0.62(7) and 0.38(7). In the final difference Fourier map, the highest peak is 0.86 Å from atom Br3 and the deepest hole is 1.05 Å from atom Br2.
Results and Discussion
Crystal Structure
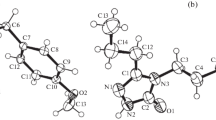
As shown in the title compound, (Fig. 1), the cyclopentane ring (C4–C9) fused to the benzene ring of the nine-membered ring system (C3–C11) adopts an envelope conformation with C11 at the tip of the envelope [the puckering parameters [19] are Q(2) = 0.330(11) Å and ϕ(2) = 141.5(19)°].

Molecular structure and atomic numbering scheme for the title compound. Displacement ellipsoids for non-H atoms are drawn at the 30% probability level. Only the major components of the disordered 1,3-dioxolane ring are shown for clarity
The C atoms of the CH2 groups of the 1,3-dioxolane ring, are disordered over two sites with an occupancy ratio of 0.62(7):0.38(7). In the disordered 1,3-dioxolane ring, its minor component adopts an envelope conformation with C3 at the tip of the envelope [the puckering parameters are Q(2) = 0.20(3) Å and ϕ(2) = 31(9)°].
In the title compound, the values of the C–Br bond lengths range from 1.915(10) to 1.972(10) Å. The relatively wide range of the Br–C–C bond angles are within the range 108.6(7)–116.2(7)° and this may indicate repulsion between the neighbouring Br atoms. In a related structure (1RS,2SR)-1,2,4,5,7-pentabromo-5-methoxyindane [9], the five Br–C distances vary from 1.884(7)° to 1.980(7)° Å. The Br–C–C angles are within the range 107.2(5)°–117.0(4)°. In trans,trans,trans-2,3,5,8-tetrabromo-1,4-dihydroxy-1,2,3,4-tetrahydronaphthalene [20], very similar values for the corresponding parameters [1.938(5)Å and 107.0(3)–121.6(4)°, respectively] were observed. All the bond lengths are within normal values (Table 1) [21].
In the crystal structure, the C–H···Br and C–H···O hydrogen bonding interactions (Table 2) help to stabilize the crystal packing of the title compound. Figure 2 shows the packing and hydrogen bonding in Table 3 of the title compound, along the a-axis.

View of the packing and hydrogen bonding of the title compound, along the a-axis. H atoms not involved in hydrogen bonding and the minor components of the disordered 1,3-dioxolane ring are omitted for the sake of clarity
Semiempirical CNDO Calculations
Theoretical calculations were carried out using the semiempirical quantum-mechanical complete neglect of differential overlap (CNDO) method [22]. The spatial view of the single molecule calculated as closed-shell in a vacuum is shown in Fig. 3 with atomic labels. When we compare the theoretical CNDO and experimental X-ray structural results with each other, it is shown that due to the intermolecular interactions in the crystal structure of the title compound, the spatial configurations obtained by the theoretical CNDO and experimental X-rays for the title compound are almost the same (see Figs. 1, 3, Table 2). We may state that the theoretical calculation (based on isolated molecules) of the title compound supports the suggestion that the present intra and intermolecular interactions in the title compound influence crystal packing. The HOMO and LUMO energy levels of the title compound are −4.1128 and 0.8545 eV, respectively. Its calculated molecule dipole moment is 8.507 Debye (1D = 3.33564 × 10−30 C.m.). The charges at atoms C10, C11, O1, O2, Br1, Br2 and Br3 are −0.3025, −0.3029, 0.0535, 0.1285, 0.2418, 0.1724 and −0.5880 e−, respectively.

The spatial view of the title molecule (I), calculated by the CNDO approximation
Supplementary Material
CCDC 917443 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_resquest/cif.
References
Mitrochkine A, Eydoux F, Martres M, Gil G, Heumann A, Reglier M (1995) Tetrahedron Asymmetry 6:59–62
Catto M, Aliano R, Carotti A, Cellamare S, Palluotto F, Purgatorio R, Stradis AD, Campagna F (2010) Eur J Med Chem 45:1359–1366
Wu YJ (2006) Tetrahedron Lett 47:8459–8461
McClure KJ, Maher M, Wu N, Chaplan SR, Erkert WA, Lee DH, Wickenden AD, Hermann M, Allison B, Hawryluk N, Breitenbucher GJ, Grice CA (2011) Bioorg Med Chem Lett 21:5197–5201
Snyder SA, Brill ZG (2011) Org Lett 13:5524–5527
Çakmak O, Erenler R, Tutar A, Çelik N (2006) J Org Chem 71:1795–1801
Erenler, R., Demirtas, I., Büyükkidan, B. , Çakmak, O. (2006). J. Chem. Res. pp. 753–757
Erenler R, Çakmak O (2004) J Chem Res 68:566–569
Akkurt M, Çelik İ, Berkil K, Tutar A, Ersanlı CC, Çakmak O, Büyükgüngör O (2005) Acta Crystallogr E 61:o475–o477
Çelik İ, Akkurt M, Yılmaz M, Tutar A, Erenler R, Kazak C (2012) Acta Crystallogr E 68:o1884
Hileman B (1993) Chem Eng News 19:11–13
Crosman A, Gelbard G, Poncelet G, Porvulescu VI (2004) Appl Catal A 264:23–32
Tutar A, Çakmak O, Balci M (2001) Tetrahedron 57:9759–9763
Rigaku/MSC (2005) Crystalclear. Rigaku/MSC, The Woodlands
Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Crystallogr 32:115
Sheldrick GM (2008) Acta Crystallogr A 64:112
Farrugia LJ (1997) J Appl Crystallogr 30:565
Spek AL (2009) Acta Crystallogr D 65:148–155
Cremer D, Pople JA (1975) J Am Chem Soc 97:1354
Akkurt M, Çelik I, Erenler R, Çakmak O, Ersanli CC, Büyükgüngör O (2004) Acta Crystallogr E 60:o2096
Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R (1987) J Chem Soc Perkin Trans 2:S1–S19
Pople JA, Beveridge DL (1970) Approximate molecular orbital theory. McGraw-Hill, New York
Acknowledgments
The author thanks Prof. Dr. A. Daştan, Atatürk University, Erzurum, for the gift sample of the title compound.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Çelik, İ. Synthesis, Crystal and Molecular Structure of 2′,3′,3′-Tribromo-2′,3′-dihydrospiro[1,3-dioxolane-2,1′-indene]. J Chem Crystallogr 43, 390–393 (2013). https://doi.org/10.1007/s10870-013-0433-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-013-0433-y